
|
|
1.IntroductionSolar energy is considered as one of the promising clean energy sources which is abundantly available (120,000 TW)1 and can be utilized to meet the increasing energy demand of the world if it is coupled with a suitable means of capturing and conversion technology. One such avenue for energy storage is photoelectrochemical cells (PECs) for the production of hydrogen. With that, the energy is stored in the form of chemical bonds and is later available on demand.2–10 PEC systems, in principle, are analogs to commercial electrolyzers, but the voltage required to electrolyze water comes directly from the photovoltage generated from absorbed solar photons. The photoabsorbing material (mostly semiconductors) should produce photovoltages sufficient to drive the water splitting reaction into hydrogen and oxygen, which necessitates band positions of the semiconducutor straddling the water reduction and oxidation potentials. Furthermore, the photoabsorbing material should be stable under solar irradiation, cheap and abundant, and should possess enough catalytic activity and selectivity. Thus, finding suitable semiconductors which satisfy these criteria poses a primary challenge for this technology to be used as an economical viable route for renewable energy. Numerous inorganic compounds from a variety of material classes have been studied in regard to their suitability as electrode materials for PEC application.2,8,10 Metal oxides are one of the promising materials for PEC water splitting. The vast amount of research on metal oxide semiconductors for PEC is driven by (1) the cost reduction arising from the abundance of metal oxides, (2) stability under PEC operating conditions, and (3) the facile synthetic procedures which do not require vacuum facilities. Spinel ferrites, with the general empirical formula, , are widely investigated as magnetic materials and used as spin filters in spintronics.11,12 They also find wide applications in the area of electrochemical energy storage (batteries and electrochemical capacitors) and photocatalysis.13–16 Spinel ferrites exhibit attractive photoelectrochemical activities originating from the (1) narrow optical bandgap () for efficiently harvesting light of the visible solar spectrum and (2) multiple oxidation states stabilized by the spinel structure with individual transition metals of known catalytic properties. Furthermore, as the constitute transition metals are abundant and are of low cost, they are promising candidates for PEC scaleup applications. In the following sections of this mini-review, we cover a brief introduction of PEC to provide an overview of the working principles and the reactions involved. Then the structural properties and some fundamental aspects of spinel ferrite materials () will be presented. Recent advances in the photoelectrochemical application of spinel ferrite for solar-assisted electroreduction or oxidation of water follow. Finally, theoretical investigations focusing on the magnetic, electronic, and optical properties of spinel ferrites will be summarized. 1.1.Basic Working Principle of Photoelectrochemical CellsPhotoelectrochemical water splitting into hydrogen and oxygen is thermodynamically an up-hill process with the free-energy barrier of or under standard conditions. The photoelectrolysis of water consists of five main steps, as depicted in Fig. 1: (1) absorption of photons with energy greater than the bandgap of the semiconductor, (2) photogeneration of electron–hole pairs, (3) band bending at the semiconductor–electrolyte interface leading to separation of charge carriers, (4) diffusion of charge carriers toward the semiconductor–electrolyte interface, and (5) redox reaction involving charge carriers and solution species (oxidation of water to and reduction of water to ). Additionally, the protons are transported across the electrolyte through proton permeable membranes and the electrons travel to the external circuit to complete the process. The overall solar splitting of water involves two half reactions taking place at the photoanode and photocathode simultaneously. For a PEC system comprising an -type photoanode and -type photocathode material, the two half reactions in alkaline media () can be expressed as follows: Three types of PEC configuration are demonstrated:9 (1) “photoanode coupled with metal electrode,” (2) “photocathode coupled with metal electrode,” and (3) “photoanode coupled with photocathode” [Fig. 1(a)]. The latter configuration allows the use of only solar photons for the overall splitting of water without using precious metal electrodes (typically, Pt for evolution and Ru and Ir oxides for evolution are used) and can operate without external bias.5,17 It can be arranged in stack or side-by-side layouts [Figs. 1(b) and 1(c)]. In a stack, the electrode with the smaller bandgap would be placed behind the electrode with the larger bandgap so that the high energy photons would be absorbed first and the lower energy photons transmitted to the back electrode. Using a combination of the two semiconductors with bandgaps of 1.84 eV for the top and 1.23 eV for the bottom electrode, a theoretical solar-to-hydrogen (STH) efficiency of 23% is predicted.17 The side-by-side configuration is preferable when the two photoelectrodes have similar bandgaps (but different band alignments each optimized for water oxidation or reduction reactions), allowing each to have access to full solar illumination. In this case, the highest STH efficiency is reduced to 16% when the bandgap for the two semiconductors is 1.59 eV. Additionally, the bandgaps have to be close to each other to absorb the same number of solar photons in order to match the photocurrents for hydrogen evolution reaction (HER) and oxygen evolution reaction (OER).18 Furthermore, this particular configuration allows the use of smaller bandgap materials which otherwise cannot be used for stacked configuration and can drive the two half reactions with a smaller photovoltage than that required for a single photoabsorber material.17 Note that in both approaches, the STH efficiency prediction takes into account the major potential losses associated with (1) free-energy loss (the difference between the acquired photovoltage and the bandgap) ranging from 0.5 to 0.6 V for highly crystalline semiconductors but expected to be higher for oxide based semiconductors, (2) kinetic overpotentials for the HER and OER (highly dependent on the choice of the catalyst, e.g., 0.05 V for Pt and 0.4 V for ),19 and (3) other potential losses including resistive and charge transport. Notably, the required bandgap is well above the thermodynamic potential of 1.23 eV and should be sufficient to split water and overcome the electrochemical overpotentials (Fig. 3). However, most real systems have higher potential losses (entropy, reflection) even under the assumption of ideal band alignments, and the minimal bandgap required to split water can be as high as 2.0 to 2.3 eV.20,21 Several excellent reviews appeared recently on topics related to solar water splitting with comprehensive coverage on the general principles of PEC and cell design and configurations, and readers are referred to these reviews for in depth discussions of the topic.5,9,22,23Fig. 1Schematic representations of (a) the band diagram of PEC in which both the photoanode and the photocathode have the required ideal band positions for spontaneous water splitting. The total photovoltage generated is large enough to split water and to overcome the electrochemical overpotentials and for the oxidation and reduction of water, respectively. Note that the energy scale is versus NHE at . The two types of PEC configurations: (b) dual stack and (c) dual side-by-side. 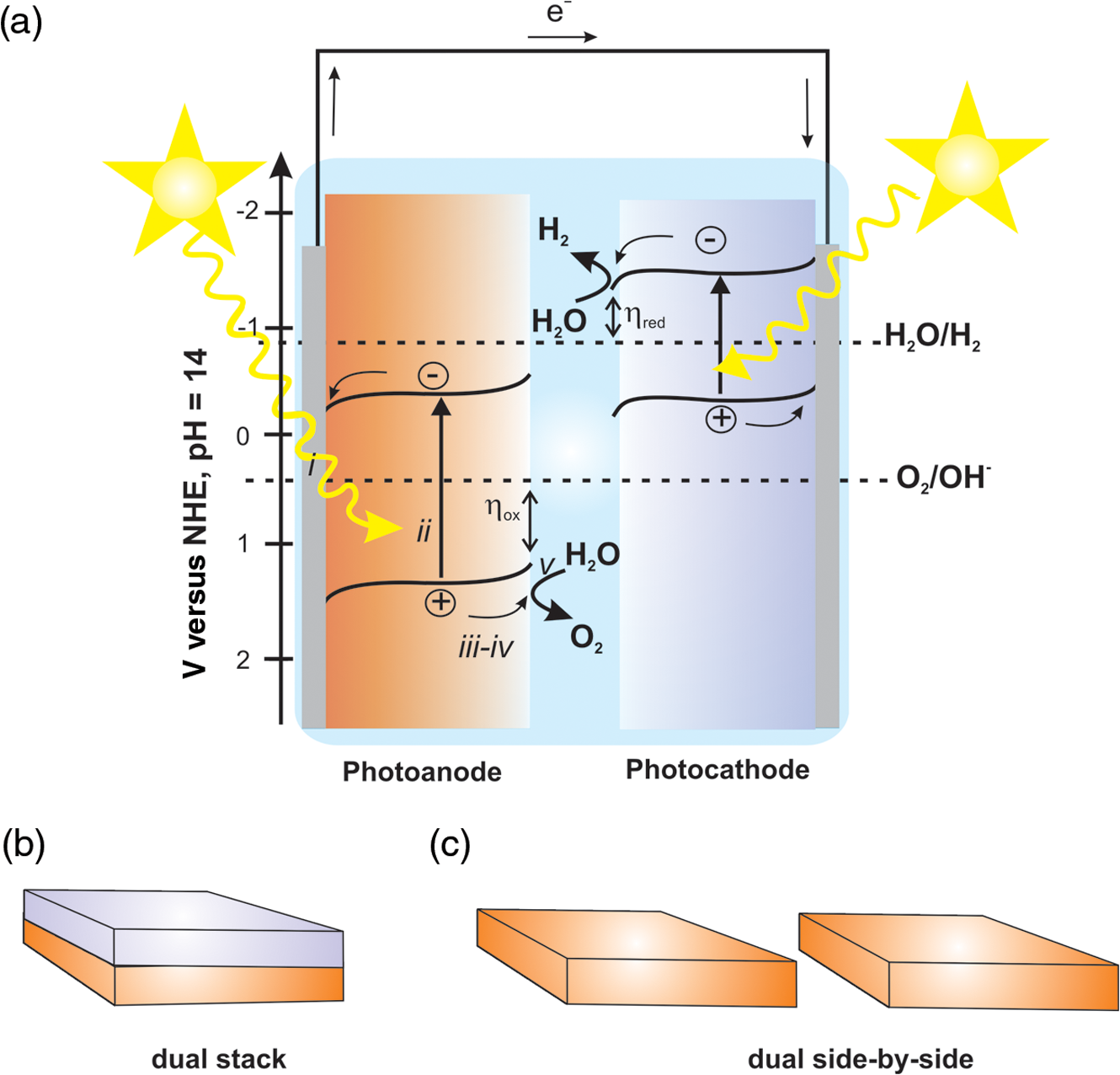 1.2.Structural Properties of Spinel FerritesSpinel ferrites are ternary transition metal oxides which are represented by a general formula , where M refers to the divalent metal ions (, Co, Zn, Ca, Mg, Mn, and so on). They are best known as magnetic materials and photocatalysts, with the most common photocatalytic application being the degradation of pollutants.12,24 The structure of spinel ferrites is derived from the mineral spinel, , by replacing the trivalent Al atom with and Mg atom by other divalent metal ions. In the spinel ferrites, the oxide anions are arranged in a cubic close-packed lattice and the cations M and Fe occupy two different crystallographic sites, namely, the tetrahedral (A) and octahedral (B) sites.25 The cubic unit cell consists of 56 atoms, 32 oxygen anions, and 24 cations, 8 of them occupying tetrahedral sites and the other 16 being located at the octahedral sites. Although the charges of M and Fe in the prototypical spinel structure () are and , respectively, other combinations are also possible. The type and the distribution of the divalent metal cations govern the final ferrite structure and dictate the chemistry of ferrites. The main factors governing the preference of the individual ions for the two crystallographic sites are the ionic radii and coordination chemistry of the ions. For example, and preferentially occupy the tetrahedral sites, whereas and have strong preference for octahedral sites. When the tetrahedral sites are occupied by divalent metal ion and the octahedral sites by , the resulting ferrites are called normal spinels (e.g., ) [Fig. 2(a)]. If the cations fully occupy the tetrahedral sites and octahedral sites are occupied evenly by and , this leads to an inverse spinel [Fig. 2(b)]; examples of these classes include but are not limited to , , and . However, there exists a certain degree of inversion in most ferrites which is determined by the fraction of ions in the octahedral sites. Fig. 2Crystal structures of spinel ferrites: (a) normal spinel, (b) inverse spinel, and (c) orthorhombic each demonstrating the three crystallographic sites. 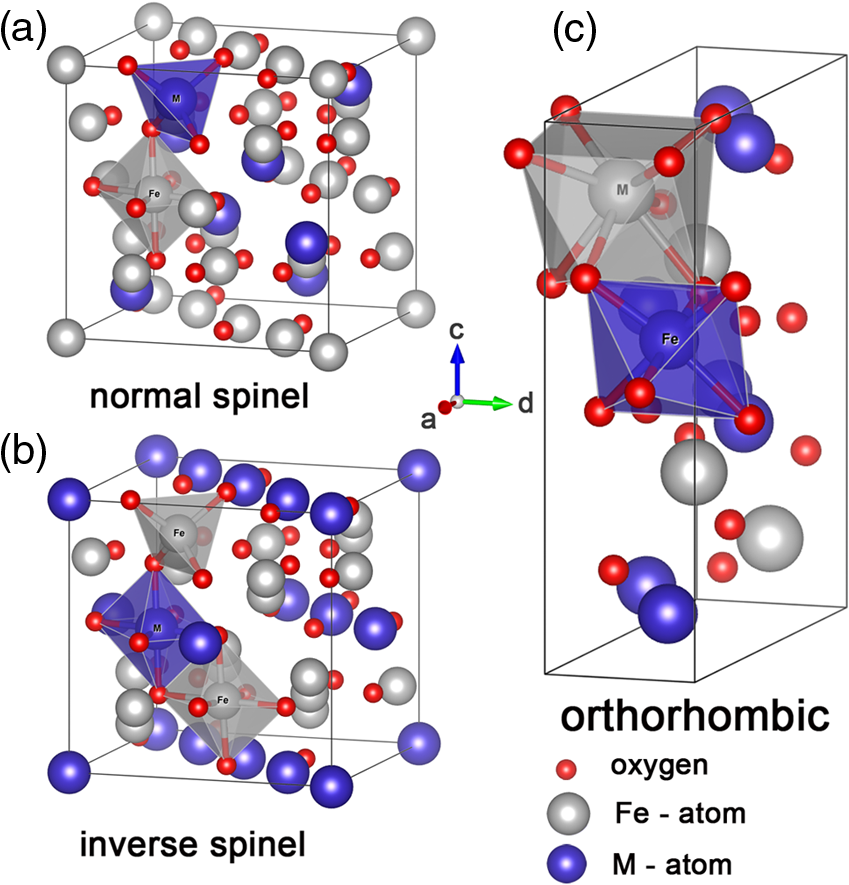 Thus, in general, the spinel ferrites can be represented by , where the superscripts A and B identify the tetrahedral and octahedral sites, respectively, and corresponds to the degree of inversion (). Mixed spinel structures are reported for Mn ferrites and Mn-Zn ferrites. Some spinel ferrites, such as , , and , are known also to form orthorhombic phases [Fig. 2(c)].26–28 Other ferrites, such as , form crystalline solids with cubic or tetragonal unit cells depending on the synthetic conditions.29 Ferrites are regarded to be chemically and thermally stable in aqueous systems.15 Considering the Pourbiax diagrams, most ferrites are stable in the alkaline or near neutral media in which most PEC investigations are carried out, however, they suffer from corrosion in acidic media.30,31 Most of them are semiconductors with bandgap energies allowing the excitation by visible light and possess energetic positions of the conduction and the valence band suitable for either reduction of protons and/or oxidation of water (Fig. 3). The theoretical and experimental bandgap energies are presented in Table 1, and the energetic positions of the valence band and the conduction band of some simple ferrites are shown in Fig. 3. The variation of , , , and in is known to affect the resistivity (conductivity),41–44 the optical properties (reflectivity), bandgap energy, and the -type behavior45,46 of the semiconductor. Also, the ability to catalyze thermal reactions is affected by the chemical nature and the magnitude of x present in an compound.47,48 Fig. 3Band positions of spinel ferrites in contact with aqueous solution referenced with NHE ( and ) relative to the standard potentials for the reduction and oxidation of water. Note that the variations of the band positions for some of the spinel ferrites data were collected from references cited in this article. 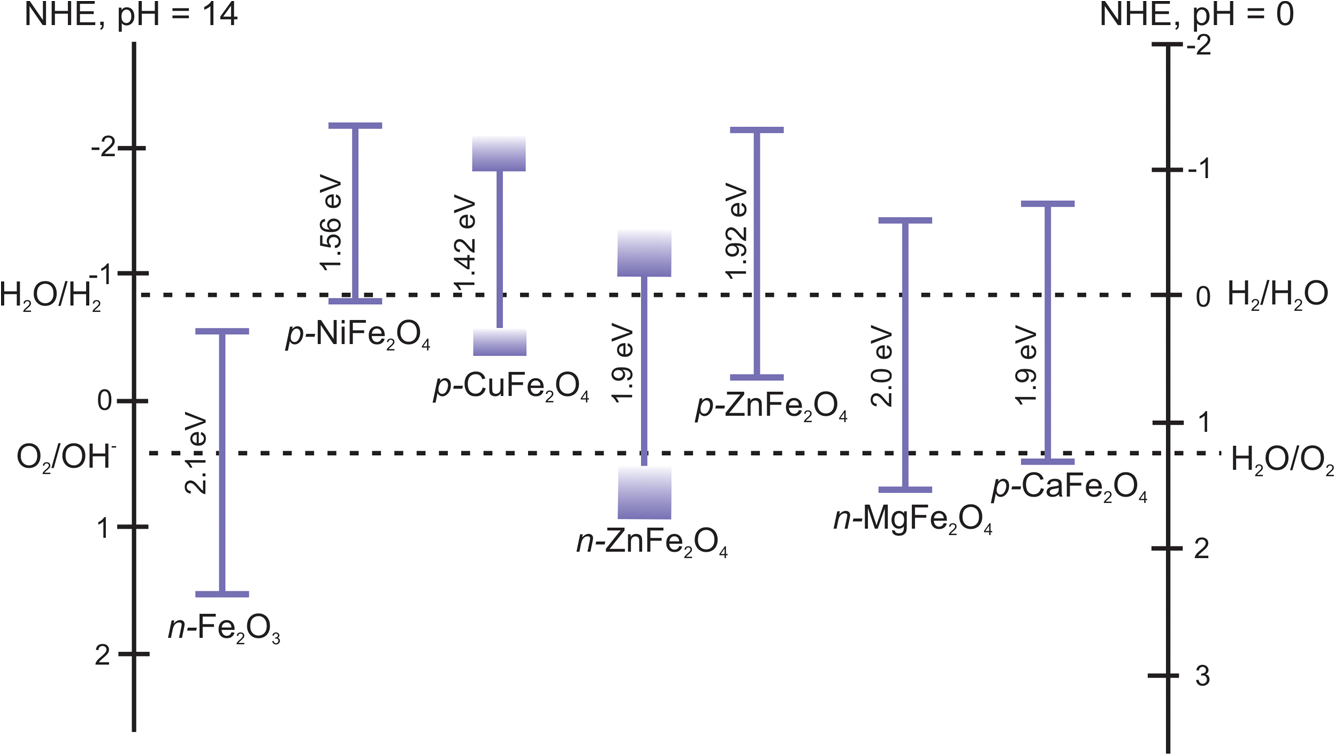 Table 1PEC performances of ferrite-based photoelectrodes.
2.Photoelectrochemical Application of Spinel FerritesOwing to the varied chemical composition, multiple valency states and choice of metal cation ferrites have attractive photoelectrochemical and catalytic activities. The improved electrical conductivity compared to the corresponding single component metal oxide (iron oxides) is mainly attributed to the presence of different metal cations which facilitate the electron transport process and/or support rich redox chemistry and also has significant importance in designing efficient photoelectrodes for PECs.49 The early works on the fundamental (photo) electrochemical investigations of ferrite electrodes date back to the late 1970s and early 1980s. Kung et al.50 reported one of the first ferrites employed for PEC, , and in the following the years, other types of ferrites were investigated including ,51 ,52,53 ,54 ,36–38,55–59 ,60 - and -type and ,45 ,39 - and -type ,40,46 ,41,51,33,61 and .41 Most of the early works focused on the basic properties of PECs, such as the determination of the flat band potentials and the energetic positions of the valance and conduction bands, however, the reported photocurrents and efficiencies were very low. Some of the fundamental semiconducting properties of these ferrites are summarized in Fig. 3 and the PEC performances are presented in Table 1. 2.1.p-Type Spinel FerritesAs with many other materials investigated for photoelectrochemical reduction of water, spinel ferrites are also studied as photocathodes for HER. In order to reduce protons to , the conduction band edge of the photocathode must be more negative than the hydrogen redox potential. Considering the band diagrams in Fig. 3, most of the spinel ferrites meet this criterion, however, only -type ,36–38,55–59 ,39 and 40 have been studied in PECs. Furthermore, the mechanism of HER is pH dependent; in acidic media, the reaction mainly involves proton reduction while the reduction of water to hydroxide ions is the primary route in alkaline solutions. Hence, PEC studies involving spinel ferrites are preferably conducted in neutral or basic solutions, as most spinel ferrites are not stable in acidic media. One of the most investigated -type ferrite is . Matsumoto et al.55 reported a -type for the photoelectrochemical reduction of water. The electrodes are prepared as pressed pellets and sintered at 1200°C followed by oxidation under at 1000°C. The photocathode exhibits -type behavior from Mott–Schottky analysis. However, the cathodic photocurrent for HER is negligibly low and when the electrode is coupled with an -type , photoelectrolysis of water without external bias results in an STH conversion efficiency of . Matsumoto49 summarized his results on ferrites and other oxide semiconductors, compiled data on the bandgap, and formulated an empirical relation between the bandgap, the conduction band edge, and the valence band edge. Cao et al.36 have investigated the visible light-induced water splitting reaction employing a photocathode. The photocathodes have been fabricated by depositing thin films on fluorine-doped tin oxide (FTO) coated glass employing a pulsed laser deposition method. A hydrogen evolution rate of was observed under visible light irradiation (300 W Xe) using a Pt metal electrode without applying any additional bias. In a three-electrode configuration, a cathodic photocurrent was observed at values more negative than . A photocurrent density of at was reported as being significantly larger than the values reported by Matsumoto et al.55,56 for metal-loaded photoelectrodes, probably due to shorter electron transfer distances in the thinner films and higher electric conductivity.36 Furthermore, Ye et al. have compared the photoelectrochemical properties of , , , and multiple junction photoelectrodes. The electrodes have been prepared by a pulsed laser deposition method using and pellets as the targets and FTO as the substrate. The authors observed a photocathodic current assigned to the reduction of water on a single-layer thin film, and a photoanodic current due to the oxidization of water on a single-layer thin film. An photoelectrode exhibited a negative photocurrent and a positive open circuit photovoltage (, , ) indicating that this electrode with a layer at the surface contacting the electrolyte acts as a photocathode. The photovoltage generated in such a system is controlled by the built-in junction potential (determined by the number of junctions) and the open circuit voltage (controlled by the type of semiconductor in contact with the electrolyte), as illustrated in Figs. 4(a) and 4(b). Investigating the photoelectrochemical properties of four multiple-junction photoelectrodes with the same single-layer thickness of 10 to 15 nm, but with an increasing number of layers (, 15, 20, and 25), has a remarkable effect on the photocurrent density and the onset potential was observed. The 20-junction photoelectrode showed the highest photocurrent density ( at ) and the most positive onset potential () of all four samples [Fig. 4(c)]. Furthermore, the 20-junction photoelectrode-based PEC exhibited a high open circuit photovoltage of up to , which was much higher than that for a cell having a single junction photoelectrode exhibiting only an open circuit photovoltage of .58 Fig. 4(a) Band structure of junction immediately after light irradiation. (b) Band structure of junction under the light irradiation after reaching steady-state conditions. In both cases, the . (c) curves of different multilayer junction electrodes (0.1 M , 500 W Xe lamp). Reprinted with permission from Ref. 52. © American Chemical Society 2013. 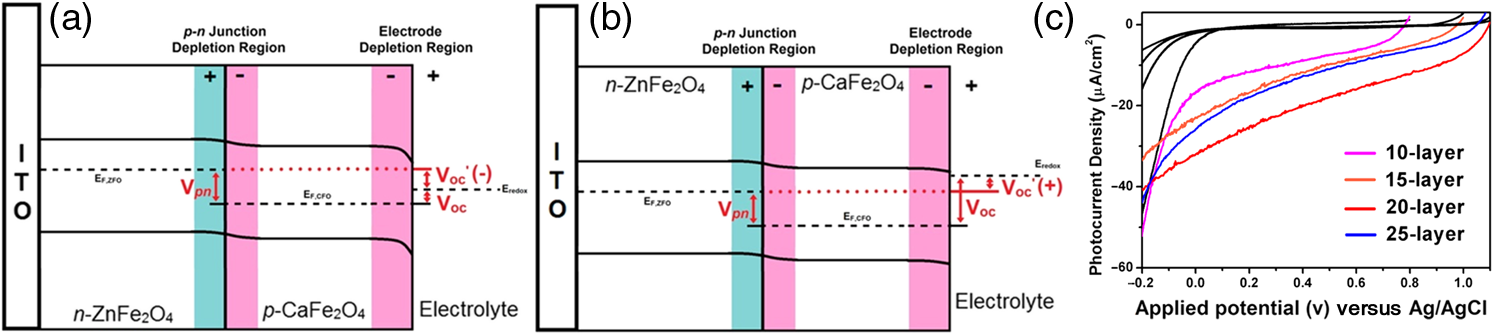 The quantum efficiency of a pristine electrode was found to be relatively low, which is presumably due to the poor mobility of the photogenerated charge carriers.56 Therefore, efforts have been made to improve the conductivity of electrodes by doping the material. Doping with Na and Mg was performed by Matsumoto et al. yielding oxides of the type . The authors suggested that as the ionic radii of and are similar to and , respectively, will substitute for and will replace . This will create acceptor levels in the bandgap leading to higher electronic conductivity; however, the reported photocurrents are still very small. The authors suggested that the formation of oxygen vacancies at higher amounts of Na () lead to the decrease of conductivity, but the formation of oxygen vacancies is not supported experimentally. In an attempt to improve the low quantum efficiency for the light-induced water splitting reaction, Sekizawa et al. have prepared various metal-doped electrodes by radio frequency magnetron cosputtering onto glass substrates coated with antimony-doped tin oxide (ATO) followed by postannealing at a low temperature. However, the doping metals were aggregated in the films after annealing as revealed by scanning transmission electron microscopy. Doping of with Au and Ag resulted in an enhancement of the photocurrent without affecting the -type conductivity. Doping with Ag resulted in an improvement of the carrier mobility together with a red-shift of the photoabsorption. Ag-doped showed a 23-fold higher photocurrent than undoped .59 It is worth mentioning that the enhanced photoresponse may originate from the dopant metals (Ag, Au, Cu/CuO) as they are known as HER cocatalysts. In a series of papers, Ida et al.37,38,57 reported the light-induced water splitting employing -based cathode coupled with different -type semiconductors. In their first report, a suspension of presynthesized powder was coated on a Pt substrate and annealed at 1100°C to 1200°C.38 The sample annealed at 1200°C resulted in a flat, well-adhered crystalline film of oriented in (320) and (420) planes. Under illumination (500 W Xe lamp), the PEC consisting of and an photoanode generated a photovoltage of 0.97 V and short-circuit photocurrent density of , respectively. The obtained photovoltage is close to the difference between the two onset potentials of (0.31 V) and (). The authors demonstrated generation of hydrogen under irradiation with visible light without applying a bias, however, the stoichiometric ratio was an order of magnitude different than the expected value of 2. The slow rate of generation is accounted for from the weak absorption of on and slow water oxidation kinetics (indicating the need for an OER catalyst). The PEC configuration, the band diagram, and the amounts of gases generated as a function of time in this study are presented in Fig. 5. Fig. 5(a) Reaction and band model in PECs using -type and -type semiconductor electrodes. (b) Current–potential curve of a photocell with () and () electrodes and model structure of measurement cell and (c) amount of hydrogen and oxygen gases generated from the photocell short-circuited by connecting the and electrodes as a function of illumination time. Measurements were carried out in 0.1 M NaOH (aq) under illumination (500 W Xe lamp). Reprinted with permission from Ref. 48. © American Chemical Society 2010. 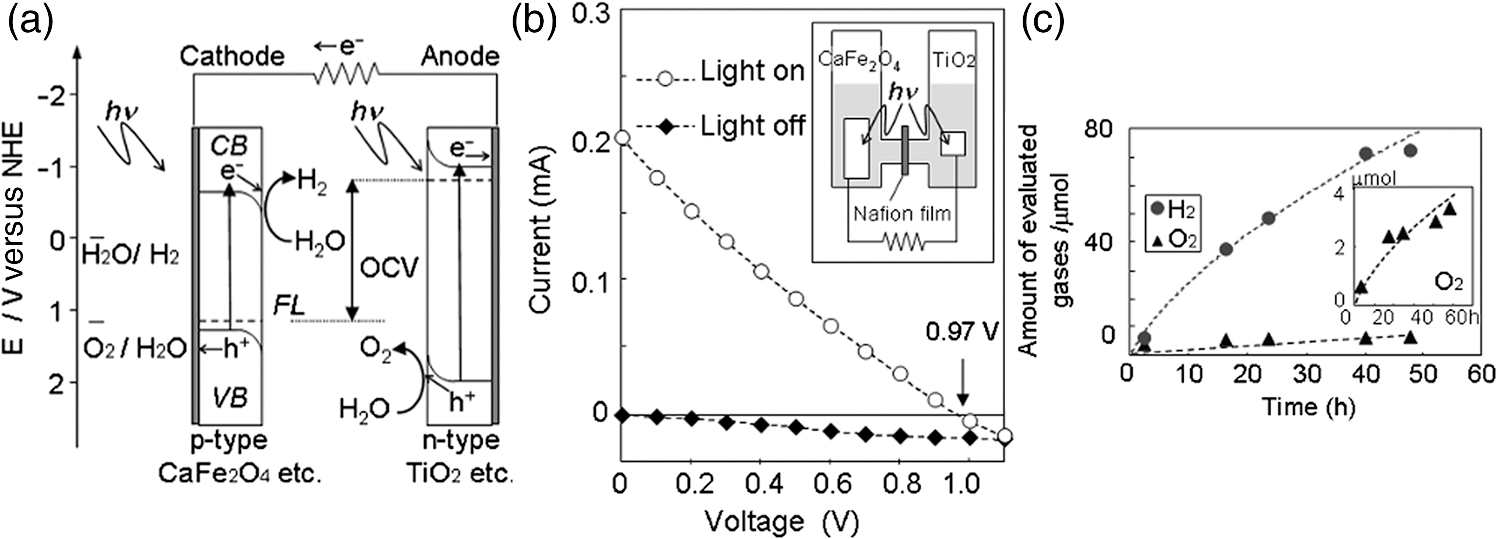 Later, the same authors reported the presence of the impurity phase in the , enhancing the short circuit photocurrent density () and slightly increasing the photovoltage (1.09 V).37 Additionally, the formation is enhanced, decreasing the ratio to 3.7, which is, however, still higher than the theoretical value of 2. -ZnO was also tested as a photoanode together with and a photovoltage of 0.82 V was generated. When employing this PEC for unbiased water splitting, only gas was detected.57 The photodissolution of -ZnO is partly responsible for the absence of gas, but there is no obvious correlation between the amount of dissolved ions and the gas evolved (which is much higher than the ion concentration detected). Very few additional spinel ferrites other than are studied as photoathodes. Yang et al. have investigated the photoelectrochemical performance of porous nanosheets on FTO. The electrodes have been prepared from an aqueous solution of Co and Fe nitrate through a template-free electrochemical deposition followed by a heat treatment at 933 K. The electrodes exhibited only a small cathodic photocurrent of in 0.1 M aqueous solution at zero bias voltage under visible light illumination (, ).39 The photoelectrochemical properties of pellets prepared by sintering sol–gel synthesized particles at 850°C were investigated by Rekhila et al. The open-circuit voltage of and short-circuit current of a two electrode configuration consisting of Pt and in a 0.5 M KCl cell were reported to be 0.43 V and under irradiation with visible light (). A photon-to-electron conversion efficiency of 0.28 was calculated. However, corrosion of the semiconductor electrode was observed under illumination as well as in the dark.40 Considering the bandgaps presented in Fig. 3, less attention is given for other -type ferrites (, , ) and more tests and investigations are still required. Sometimes, even the fundamental optical properties are controversial. For example, most report the optical indirect bandgap of as 1.3 to 1.4 eV,62,63 but Xiong et al. reported a bandgap much lower than these values (0.8 eV).64 In the authors’ opinion, one of the main reasons is that these -type ferrites are known to exist in a completely or partially inverted spinel structure.24,63 Such a degree of inversion should be well controlled and require a rational design of synthetic strategy to accurately determine the optical properties and to enhance the PEC performance. In addition, they are prone to photocorrosion particularly in acidic media, which can be alleviated by applying suitable protective layers (, ). 2.2.n-Type Spinel FerritesThe photoelectrochemical oxidation of water to requires an -type semiconductor with the valence band located more positive than the oxidation potential (1.23 V versus NHE). In contact with an aqueous electrolyte, such a semiconductor results in an upward band bending which drives the holes toward the surface leading to the oxidation of water to . Additionally, good electrical properties and stability under water oxidation conditions are needed. Among the spinel ferrites, is the promising candidate and is the only -type photoanode material reported for PEC application. Systematic investigation of the photoelectrochemical activity was reported by Tahir and Wijayantha33 and Tahir et al.61 The electrodes were prepared by aerosol-assisted chemical vapor deposition (AACVD) of alcoholic solutions of a bimetallic precursor () on FTO. The thickness, morphology, and nanostructure of the electrode were controlled by altering the solvent for dissolution of the bimetallic precursor and physical deposition parameters.33,61 The photocurrents were found to be dependent on the solvent, as well as on the deposition temperature and the deposition time. A maximum photocurrent density of at 1.23 V versus RHE was obtained with a electrode synthesized using a 0.1 M solution of the bimetallic precursor in ethanol, the optimum deposition temperature of 450°C, and a deposition time of 35 min. This electrode showed an incident-photon-to-electron conversion efficiency of 13.5% at 350 nm and an applied potential of 1.23 V versus RHE.33 In the AACVD process, the aerosol droplet size (controlled by the solvent) and the enthalpy of combustion determine the decomposition pathway (homogeneous versus heterogeneous), thus varying the methanol/ethanol ratio of the solvent resulted in a change in the texture of the electrode. A compact film composed of hexagonal like particles was obtained in pure methanol, but the structure transformed in to porous nanorod films when ethanol was used as the solvent. Intermediate structures were obtained by varying the methanol/ethanol ratio. The textured electrodes exhibited a significantly higher photocurrent under AM1.5 illumination compared to their compact counterparts. The authors attributed this behavior to the improved collection of the photogenerated minority carriers at the interface as the average feature size gradually decreased from (methanol) to (ethanol).61 In general, ferrites as photoelectrodes need high temperatures to crystallize (). This limits the choice of support materials and poses a critical challenge to maintain the desired electrode material properties such as surface area and porosity. Recently, Kim et al. introduced a hybrid microwave annealing (HMA) postsynthetic heat treatment with graphite powder as the susceptor being compatible to most transparent conducting glasses. They treated solution processed -FeOOH nanorods with a Zn nitrate solution and obtained nanorods after thermal treatment at 550°C for 3 h. Some unwanted ZnO on the nanorods was removed in NaOH. Subsequently, the nanorods were subjected to a second heating step at 800°C (20 min) or to HMA (5 min) to increase the crystallinity. The HMA-treated nanorods exhibited at 1.23 V versus RHE (1 M NaOH) and AM 1.5G illumination a photocurrent of , which was a 10- to 15-fold increase in comparison to conventional thermally treated electrodes and was stable for at least 3 h. The authors claimed that stoichiometric amounts of and can be measured with Faradaic efficiencies (= actual gas evolution rate/rate expected from current) of 90% to 100%. The improved performance after the HMA treatment was attributed to better crystallinity and reduced surface defects as evinced by electrochemical impedance spectroscopy.65 Extending their work, the same authors recently reported the influence of the composition of the annealing atmosphere on the photoelectrochemical behavior of (Fig. 6).34 nanorod films were first treated at 800°C (20 min in air) followed by a mild temperature treatment at 200°C (2 h) either under vacuum, air, or hydrogen atmosphere. The hydrogen and vacuum post-thermal treatment enhanced the photoactivity about 20-fold [Fig. 6(b)]. The increased activity is attributed to oxygen vacancies created in the lattice due to the limited oxidation environment as proven by O 1s XPS. Optimal oxygen vacancy concentrations increase the majority carrier density and lead to improved charge separation. The highest photocurrent was observed for the hydrogen treated sample, at 1.23 V versus RHE under AM 1.5G illumination. The authors suggested two types of mechanisms of how the lattice O () is replaced by oxygen vacancies () under controlled hydrogen and vacuum atmosphere [Fig. 6(a)]. In hydrogen atmosphere, the reacts with and leaves the lattice as water molecules: in vacuum, the oxygen removal is represented by Eq. (5) and the concentration of the oxygen vacancies is given in Eq. (6): where represents the electron density and is the partial pressure of the oxygen.Fig. 6(a) Schematic representation of different mechanisms of generating oxygen vacancies by post-treatments under hydrogen or vacuum conditions, (b) characteristics of photoanodes before and after post-treatment [hydrogen (H), vacuum (V), and atmospheric air (A)] under AM 1.5G illumination () in 1 M NaOH electrolyte (). Reprinted with permission from Ref. 60. © Royal Society of Chemistry 2015.  In general, for most iron based -type semiconductor oxide materials, the diffusion length of the minority carrier is very short leading to an inherently high charge recombination rate, which limits the efficiency of the PECs. Several strategies to address this issue were developed including nanostructuring33,61,65 and doping.59 Another attractive nanostructuring strategy is to use a structured transparent conductive oxide current collector to capture and tunnel the photogenerated electrons readily while the large interfacial area allows efficient transfer of the holes to the solution. This strategy was recently demonstrated for decorated Al-doped ZnO (AZO) nanowire films.32 The Al:ZnO nanowires were grown on FTO substrate hydrothermally at 88°C and treated with an ethanolic solution of . Subsequent annealing at 550°C leads to -coated Al:ZnO nanowires [Figs. 7(a)–7(d)]. Depending on the time of exposure, the nanowires can be converted to nanotubes due to the dissolution of Al:ZnO in acidic solution. The photoanode shows outstanding photoelectrochemical performance with low onset potential (0.38 V versus RHE) with a photocurrent density of at 1.23 V versus RHE [Fig. 7(e)]. The synergy of high conductivity of Al:ZnO, the nanowire morphology for charge separation, and the visible light absorption of coating are attributed to be responsible for the high photoelectrochemical performance. Fig. 7SEM images of (a) 0.5% AZO and 0.5% AZO–ZFO with treatment times of (b) 1 min, (c) 3 min, (d) 7 min, (e) LSV plots of 0.5% AZO and 0.5% AZO–ZFO photoanodes under chopped illumination and (e) the corresponding ABPE plots. Insets show the schematic illustration of morphology evolution for the AZO and AZO–ZFO composite photoanodes with different treatment times. Reprinted with permission from Ref. 61. © Royal Society of Chemistry 2016. 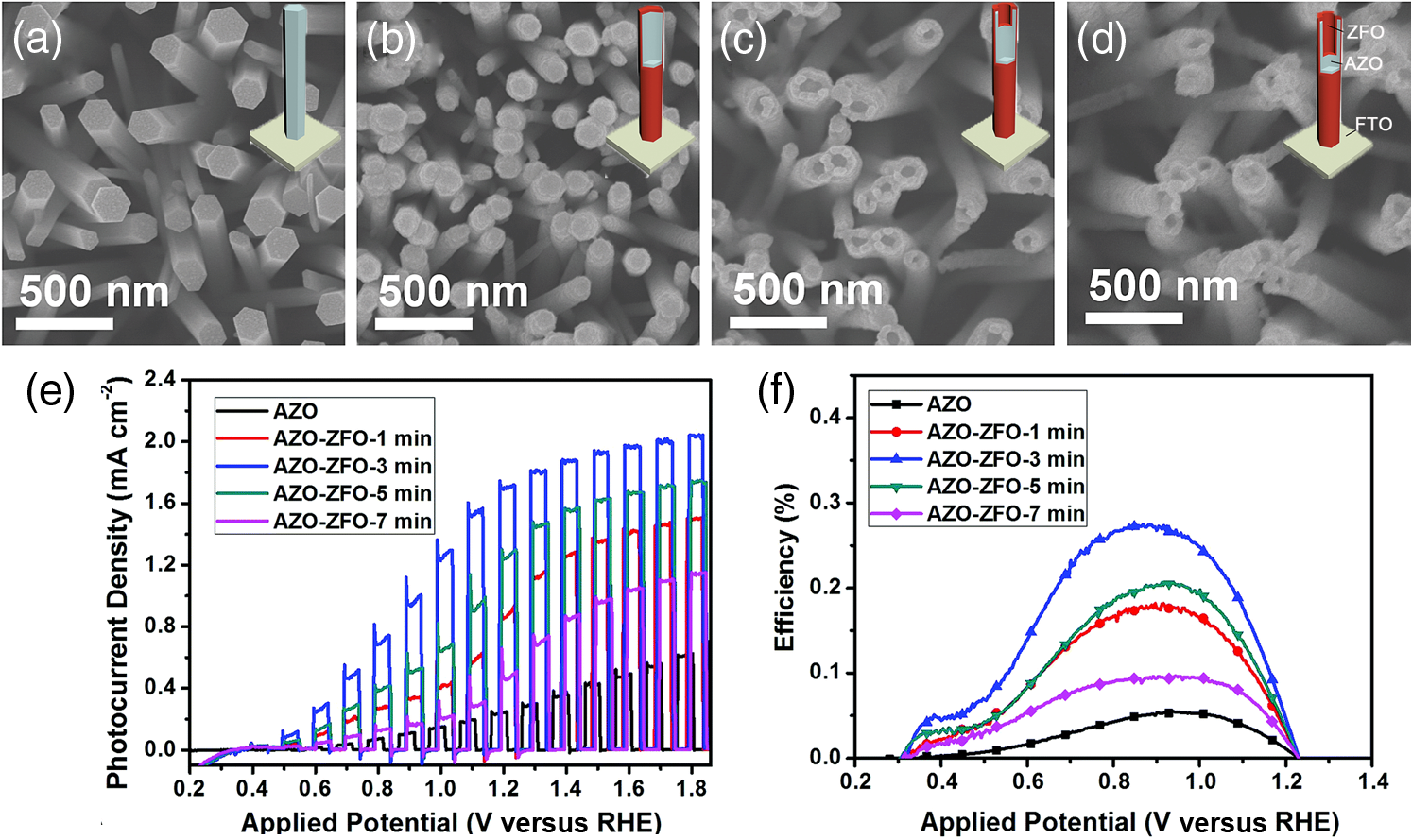 Recently, Hufnagel et al.35 prepared mesoporous thin films on a macroporous ATO scaffold using atomic layer deposition (ALD). The photoresponse of the electrodes was tested in three electrode PECs and the electrodes exhibited 4- to 5-fold higher photocurrent density ( at 1.23 versus RHE) compared to nonstructured films prepared in a similar way ( at 1.23 versus RHE). Additionally, the authors show that such electrodes have more negative photocurrent onsets (0.9 V versus RHE) compared to reported values.33 Spinel ferrites were recently investigated for construction of heterojunction electrodes to improve the photoelectrochemical performance of other widely used semiconductors. In this regard, heterojunction electrodes such as ,66–68 ,69,70 ,71 and 72 as photoanodes for the OER were studied. Table 2 presents the PEC performance of ferrite heterojunction photoelectrodes. Borse et al. prepared layers on stainless steel by depositing an aqueous solution of Zn and Fe salts employing a plasma spray method and investigated the photoelectrochemical activity of the electrode. Under simulated solar light (AM1.5G, ) with a bias of 1.4 V versus RHE, a photocurrent of is measured, which is fivefold higher than for pristine . The authors also reported hydrogen production in a two electrode set-up employing graphite as the counter electrode. Again, the composite photoanode exhibited a significantly higher photoactivity than a bare photoelectrode. The rates of HER at the and the photoanode were calculated to be 46.3 and , respectively, resulting in STH conversion efficiencies of 0.06 and 0.0125, respectively. However, no data were given for the formation of molecular oxygen. The results of electrochemical impedance spectroscopy evinced a significantly lower interfacial charge transfer resistance of the composite electrode than the electrode.67 Table 2PEC performance of ferrite based composite photoelectrodes.
Note: EPD, electrophoretic deposition. nanorod composite photoanodes have also been prepared using hydrothermally grown FeOOH nanorods and subsequent treatment with different concentrations of Zn precursor. After calcinations at 750°C, a composite electrode was obtained.68 The photocurrent density for the composite electrode was at versus RHE, which was almost twice as high as that for a electrode (). McDonald et al. employed the electrodeposition route and obtained photoelectrodes composed of an (hematite) core and a shell as confirmed by XRD.66 The electrodeposited -FeOOH films on FTO were converted into by heat treatment and subsequent treatment with Zn-containing solution on top of film yielded a Zn-rich top layer on after annealing. The highest photocurrent was obtained with a composite electrode exhibiting a ratio of 1. The increase in the photocurrent of the heterojunction electrodes compared to the bare electrode was explained as being due to the enhanced electron hole separation at the interface. A further enhancement in photocurrent was obtained by a treatment of the composite electrodes with an solution yielding thin layers of a solid solution ( or ) after heat treatment. With this, the number of surface states that serve as electron–hole recombination centers is probably reduced. But it was also observed that both the formation of a layer and the incorporation of into the surface made the surface less catalytic for the OER. However, when was introduced into the surface of the composite electrodes as oxygen evolution catalysts, the onset of the photocurrent was shifted to more negative voltage and the overall photocurrent was improved.66 Furthermore, heterostructures are also prepared using a combination of solution-phase materials growth techniques and ALD.73 The nanowires were hydrothermally grown on FTO substrate and later infiltrated by Zn precursor using ALD. The composite electrodes show an enhanced visible light photoresponse compared to bare photoelectrodes. The authors suggest that the extended visible light absorption by the , the nanowire morphology of the , and favorable band edge positions are the main reasons for the increased photoresponse. A related photoanode has been prepared by anisotropic growth of a -FeOOH film on FTO from an aqueous solution containing Fe and Ca ions followed by two-step thermal annealing at 550°C and 800°C. The authors suggested that this procedure induces the formation of a heterojunction photoanode. The presence of Ca in the film, leading to the formation of , has been proven by XPS measurements. Under illumination (AM 1.5G, ), the heterojunction photoanode exhibits a photocurrent density of at 1.23 V versus RHE, which is a 100% higher photocurrent response than that obtained using a bare electrode. Based on electrochemical impedance spectroscopy, the photocurrent enhancement has again been attributed to an enhanced charge carrier separation and a reduced resistance of the interfacial charge transfer between the electrolyte and the electrode.69 Kim et al.71,74 reported the preparation and characterization of modified TaON and heterojunction photoanodes. Both -type semiconductors are known to be suitable anode materials for solar-driven water splitting in PECs. The valence band of is more positive than the water oxidation potential, and both semiconductors TaON and form staggered relative band positions with the ferrite as required for an effective heterojunction photoanode. In both cases, , which has been synthesized by a conventional solid state reaction, was deposited on top of the -semiconductor/FTO electrode by electrophoresis. The pristine TaON electrode showed an anodic photocurrent density of at 1.23 V versus RHE (0.5 M NaOH, ). The layer on the surface of a TaON electrode resulted in a significant increase of the photocurrent density (). The observed photocurrent was found to be a result of overall water splitting yielding and in a ratio of 1.5, accompanied, however, by a deterioration of the TaON. Impedance spectroscopic analysis indicated that the formation of the heterojunction increased the photocurrent density by reducing the resistance of the charge carrier transport and, consequently, enhancing the electron–hole separation.71 Anodic photocurrents have likewise been observed for both and electrodes (0.5 M , AM 1.5G ); the bare electrode showed a photocurrent density of at 1.23 V versus RHE V while the heterojunction photoanode exhibited , comprising an increase of 65% over that measured at the electrode. The formation of the heterojunction was found to reduce the recombination of the photogenerated charge carriers on the electrode surface with little effect on bulk recombination as evinced by an investigation of the interfacial transfer of charge carriers using hydrogen peroxide as an electron donor.74 In the above demonstrated heterojunction electrodes, efficient charge separation is the key factor for improved photoelectrochemical activity. This is demonstrated in the energy diagrams of the composite electrodes in Fig. 8. The ferrites ( and ) have valence band energies located between the water oxidation potential and that of the valence band of the second semiconductor. Thus, holes generated at the more positive valence band are extracted to the ferrite valence band reducing bulk recombination and allowing successful injection of the holes to the electrolyte. On the other hand, as the conduction band of the ferrites is situated at a more negative potential, the photogenerated electrons will be easily transferred to the conduction band of the second semiconductor for collection at the back contact. Fig. 8Schematic representation of the band positions of heterojunction electrodes showing the flow of photogenertaed charge carriers: (a) the /TaON/ ( junction) and (b) the ( junction). 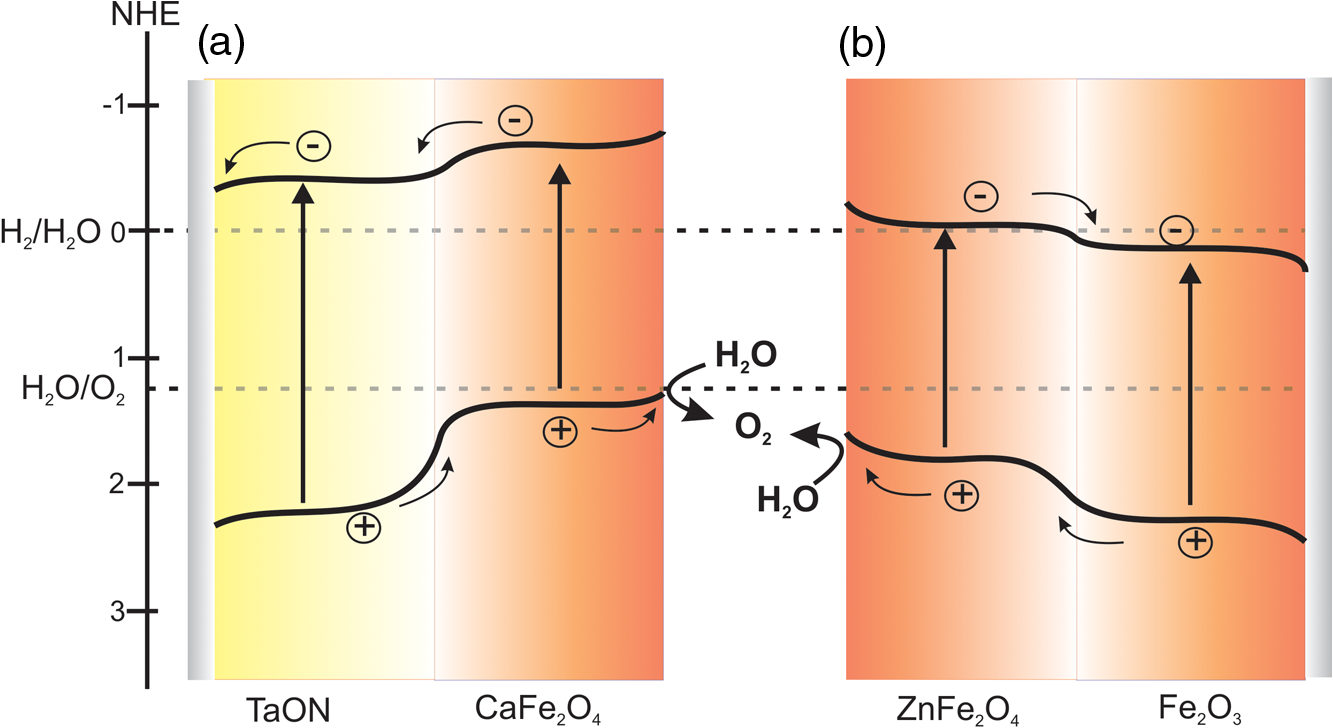 Furthermore, the modification of the heterojunction photoanodes by depositing OER cocatalysts results in higher photocurrent densities by decreasing the onset potential and facilitating the interfacial charge transfer. In this regard, “cobalt phosphate” (CoPi) is used widely as an OER catalyst.75–78 For example, in the 71 heterojunction electrode system, after deposition of CoPi and with an applied bias of 1.23 V versus RHE, and were generated with nearly of stoichiometric ratio of 2.1 ( and were produced within 3 h of illumination with ). The STH efficiency was 0.053% at 1.0 V versus RHE, but reached 0.55% when a PV device is coupled in tandem configuration (assuming the applied voltage is zero). However, the initial current decreased within 3 h to about 50%. The beneficial role of the CoPi cocatalyst was revealed by performing the gas evolution experiment with a photoanode in the absence of CoPi. Under these experimental conditions, no constant photocurrent was obtained, the faradaic efficiencies decreased (50% to 70%), and the ratio became less than stoichiometric (1.51) due to the self-oxidation of TaON.71 Similar results have been obtained with composite photoanodes. In this case, the CoPi modified electrode exhibited a lower photocurrent density when compared to the heterojunction electrode, but an improved stability of the current density was observed indicating that the presence of the OER cocatalyst is beneficial for the stabilization of the heterojunction. The evolution of and during the photoelectrochemical water splitting reaction was measured in a three electrode set-up in phosphate buffer () under applied bias. The total amounts of and evolved within 2 h of illumination with visible light () were 297 and , respectively. The resulting ratio of 2.1 confirmed that the generation of the photocurrent was mainly due to the water splitting reaction. The Faradic efficiency during this reaction was reported to be about 78% to 88%. The photocurrent density was initially , but dropped rapidly during the first 30 min of illumination and decreased slowly afterward.74 In general, the above few examples of spinel ferrite-based PECs demonstrated the attractive photoelectrochemical properties of these materials. The measured photocurrents of PECs with ferrite photoelectrodes are still low, but different strategies proved that higher photocurrents can be achieved. The main contributors for low photoelectrochemical performances are identified as (1) slow interfacial charge carrier transfer, (2) inherently high charge carrier recombination rates, and (3) loss of interfacial area due to the high thermal treatment. Strategies to address these bottle necks, including (1) nanostructuring, (2) forming heterojunction structures, (3) cocatalyst coating, and (4) control of the defect chemistry are shown to enhance the photoelectrochemical performance. It is worth mentioning that the reported experimental band positions of some of the spinel ferrites related to HER and OER potentials are still arguable. Based on the energetic band diagrams of ferrites presented in Fig. 3, and are, in principle, capable of producing hydrogen. However, to the best of the authors’ knowledge, there is no literature report proving this. For example, the reported flatband potentials of (0.6 to 0.8 V versus RHE)33,35 suggest the conduction band lies slightly below the HER potential. We also found that the conduction band edge is slightly positive () to that of the proton reduction potential, but we are still in the process of verifying this. Thus, more experimental investigations are required to refine the existing photoelectrochemical data. Furthermore, is also interesting for OER, but there are no reports except the fundamental investigation reported by Benco and Koffyberg.52 However, there are a few reports on photocatalytic production using which to some extent show the potential of this material.53,72 3.Theoretical InvestigationsConsidering the above-mentioned PEC applications of spinel ferrites, several questions concerning the water splitting process arise that cannot be easily answered. Theoretical investigations allow an insight to material on an atomistic level that the experiment cannot provide in general. For example, theory allows one to study water and hydroxyl adsorption on different sites of differently terminated ferrite surfaces, and its effect on the degree of inversion, the magnetic structure, the bandgaps, and band positions. It is well known that the accuracy of theoretical predictions of electronic properties strongly depends on the choice of the method. During the last years, several quantum chemical protocols have been developed which go beyond the traditional independent-particle model at density-functional theory (DFT) level, the most refined being self-consistently vertex corrected GW.79 A considerable number of theoretical investigations were devoted to the electronic, magnetic, structural, and energetic properties of ferrites. With a few exceptions, the quantum-chemical studies were performed at DFT level, in most cases employing the local (spin) density approximation (LSDA) or the generalized-gradient approximation (GGA). As is well known, the electronic properties of transition metal oxides are not accurately described within the LSDA or GGA, thus hybrid approaches combining DFT with unrestricted Hartree–Fock theory (HFT) are preferable. Global DFT-HFT hybrids have been demonstrated to provide accurate lattice constants, atomization energies, and bandgaps for a wide range of compounds80 but are computationally much more demanding than GGA methods. Therefore, in most studies, a semiempirical on-site correction, the so-called LSDA+U or GGA+U approach,81 is applied. In this method, the effective parameter U replaces one-center exchange and Coulomb integrals from unrestricted HFT. In principle, the value of U for each transition metal can be derived from exact theory, but in practical calculations, it is usually treated as an empirical parameter to adjust certain properties to experimental reference data. Absolute band positions thus, the work functions and fundamental bandgaps can only be obtained from two-dimensional slab model calculations of surfaces. However, recently, very accurate results for both properties were obtained for binary systems (e.g., ZnO, CdO, GaAs, GaP, InP) from self-consistent GW calculations.79 So far, most theoretical investigations of ferrite surfaces, however, have focused on the adsorption of water and other small adsorbates. The following sections provide a survey of recent theoretical studies on bulk and surface properties of ferrites. 3.1.Electronic Structure of the BulkThe effect of charge ordering in the octahedral sites of and on their electronic structure was investigated using DFT+U.82 A precise description of charge ordering was found to be crucial in determining the bandgaps of the compounds. GGA+U calculations of the electronic structure of antiferromagnetic yield an indirect bandgap of .83 The ionicity of has been determined using DFT calculations.84 Furthermore, a new developed quantum mechanical estimation method for the ionicity of spinel ferrites has been proposed and tested. On the basis of this, the ionicities of the spinel ferrites (M: Co, Cu, Fe, Mn, Ni) were calculated. The electronic structure of has been investigated using LSDA+U and hybrid-DFT in 2012.85 According to the theoretical results, the system is an indirect gap material in one of the minority channels and slightly larger direct bandgaps can be found both in the minority and majority channels. The electronic structure of (M: Ca, Mg, Zn) was investigated in a combined experimental and theoretical study.86 The DFT calculations reveal that the M-ion controllably affects the density of states of the Fe d-orbitals near the Fermi level. The electronic structure of was studied using GGA+U.87 Taking the effect of spin arrangement on symmetry into account, was classified as a semiconductor. The impact of cation distribution in on electronic structure and magnetic properties has been investigated by Feng et al.88 The lattice structure was optimized on the GGA level and the electronic structure was calculated with GGA+U. The calculated density of states shows that the distribution of Cu ions significantly impacts the electronic structure. Multilayer bispinel composites, in which one member is and the other is (M: Co, Mg, Mn, Ni), were modeled using GGA+U by Wells et al.89 It was found that substitution of the transition metal sites in the supercell produces cation charge transfers and magnetization modulation. Band shifts and gap modulation were comparable to the chemically similar bulk compounds. Two different distributions for the octahedral-site cations in and have been investigated using LDA and GGA, as well as LDA+U and GGA+U.90 It was shown that a different octahedral-site distribution impacts the density of states as well as the bandgaps in both the normal and inverse spinel configurations of these compounds. Magnetic properties and the electronic structure of have been studied using hybrid-DFT.91 The calculated density of states suggests that is an insulator. The electronic structure of normal and inverse spinel ferrites (M: Co, Fe, Mn, Ni) was investigated by self-interaction corrected LSDA.92 For both structures, all studied compounds were found to be insulating but with smaller gaps in the normal spinel structure. The calculated spin magnetic moments and exchange splitting of the conduction bands were dramatically increased when moving from the inverse spinel structure to the normal spinel. A first principle investigation of the electronic structure of (M: Co, Fe, Mn, Ni) compares the performance of LSDA and LSDA+U.93 For the LSDA+U approach, the charge ordering is stable in contrast to a metallic state given by the LSDA approach. Calculated x-ray absorption spectra as well as the x-ray magnetic circular dichroism spectra were in good agreement with the experiment. The electrical and magnetic properties of the normal and inverse spinel structures of were calculated with DFT by Zuo and Vittoria.94 The calculated bandgap suggests that is a complex insulator, in contrast to earlier LSDA and GGA calculations which suggest a half-metallic behavior. has been investigated theoretically at DFT level.95 The calculated band structure shows a low carrier density half-metal in the fully ordered state, in contrast to experimental characterizations. The computations yield a strong coupling of the energy bands at the Fermi energy to the internal structural parameter as well as strong effects on the electronic structure upon partial interchange of Fe and Mn atoms. Calculations of the K-edge x-ray absorption near-edge structure (XANES) in elemental iron and (M: Mg, Mn, Ni, Zn) were carried out by Safontseva and Nikiforov.96 It was shown that the Fe K-edge energy shift found experimentally occurs upon the transition from elemental iron to the spinel ferrites. This shift was demonstrated to be identically directed for ferrites with a normal and inverted spinel structure. A computational study of ferrimagnetic compounds using the pseudofunction method was carried out in 1996.97 Substitution of Ni with Zn enhances the localization of the 3d states of Fe on the octahedral sites, so that the O 2p-Fe 3d hybridized states can be resolved into two distinct twofold and threefold features. Normal and inverse was investigated with HF level of theory in 1996.98 From Mulliken population analysis and net spin density distributions, it was concluded that the charge states of Mn and Fe in the ground state show no evidence of charge transfer leading to at A sites and at B sites in the inverse spinel structure . An early computational study of the band structure and magnetic moments of ferrites (M: Co, Fe, Mn, Ni, Zn) on LSDA level99 only covered the metallic, high-temperature phase in the case of , Fe, Mn, Zn. In contrast, was described as an insulator. Table 3 compares experimental bandgaps of selected ferrites with calculated values gained by different theoretical protocols. It is obvious that plain DFT without any further corrections systematically underestimates the bandgap of the considered systems. The theory states that using a self-interaction corrected or a hybrid DFT approach reduces the error. Using a DFT+U framework provides results that show good agreement with the experimental data. This is easily explained by the added potential U, which is an empirical parameter that can be explicitly chosen to fit the experimental bandgap. There is no universal choice for the U parameter. Table 3Comparison of experimental and calculated bandgap energies of some selected MFe2O4 spinel ferrites.
3.2.Magnetic PropertiesSince the magnetic structure of ferrites (strongly) affects the calculated bandgap,92 the magnetic properties also have to be taken into account when discussing electronic properties. The magnetic properties of (M: Co, Fe, Mn, Ni, Zn) were studied with DFT methods.118 The theoretically obtained magnetizations were consistent with experimental results in the absence of an external field. GGA+U was applied to investigate the electronic structure and magnetic properties of .117 The calculations account for a cubic structure with ordered spins and insulating behavior. It was found that the high-spin state is favorable for the two cations Mn and Fe. The position of magnesium ions in -doped lithium ferrite of the composition has been investigated by interatomic potential and DFT calculations.119 The lowest energy structure was found for ions evenly replacing and ions on octahedral sites. This occupation affects a decrease in magnetization for the -doped ferrite relative to the undoped lithium ferrite. A computational study of the spinel ferrites and shows that LSDA+U and GGA+U allow for a good quantitative description of these materials.110 The effect of epitaxial strain on the magnetocrystalline anisotropy was investigated and the results are in good agreement with experimental observations. The structure of partially inverse spinel as well as its electronic and magnetic properties has been investigated by the GGA+U approach.120 It was found that the Co and Fe ions prefer their high-spin configurations with higher spin moments at octahedral sites. Certain investigated inversion degrees show half-metallic behavior. 3.3.Structural and Thermodynamic PropertiesA computational study of the inversion thermodynamics and electronic structure of (thio) spinels (M: Co, Cr, Mn, Ni; X: O, S) was published in 2015.121 The analysis of the configurational free energies shows that and are fully normal, and are intermediate, and and are fully inverted. The calculations illustrate that , , , and are half metals in the ferrimagnetic state when Fe is in tetrahedral positions. When M is filling the tetrahedral positions, the Cr-containing compounds and are shown to be half-metallic systems, whereas the Co and Ni spinels are shown to be insulators. Yao et al.116 investigated the structure and electronic properties of normal spinel using GGA and LDA. They suggest that the GGA functional RPBE combined with ultrasoft pseudopotentials is a good method for predicting the crystal structure of the compound. The computational results indicate that is a direct gap semiconductor and that there is a strong hybridization between the Fe 3d states and the O 2p states as well as between the Zn 3d states and O 2p states. DFT calculations at the GGA+U level were performed on (A: Fe, Ni, Zn; B: Fe, Cr) spinel oxides in order to determine thermodynamic properties.101 Calculated mixing energies quantitatively reproduce experimental data. Reactions leading to an excess of A or B, respectively, were found to be slightly exothermic in a number of spinel compounds. A set of effective chemical potentials (ECPs) that connect energies of (M: Co, Fe, Ni, Zn) spinels and oxides calculated at 0 K from DFT to free energies at high temperature and pressure in the presence of water was derived and tested.109 The ECPs were used to calculate free energies of low index stoichiometric surfaces of nickel ferrite in water, predicting surface denuding at high temperatures. A computational study compares the performance of GGA-DFT and hybrid-DFT (B3LYP) for the equilibrium structure of .104 The ground state calculated by GGA-DFT is metallic with Fd-3m symmetry while the hybrid level of theory yields a charge ordered semiconducting state with P2/c symmetry. Phonon frequency calculations showed that charge ordering causes symmetry breaking of force constants on symmetry lowering from the cubic unit cell to the monoclinic unit cell. 3.4.Surfaces and AdsorptionThe structure, electronic properties, and energetics of the surface and its interaction with water both in the absence and in the presence of surface oxygen vacancies have been studied using DFT+U.122 It was shown that water adsorbs dissociatively on the surface oxygen vacancies leading to the formation of surface hydroxyls. Furthermore, it was found that at high temperature, water desorbs leaving a surface containing oxygen vacancies. The reactivity of the surface has been studied using DFT+U.123 The surface reactivity is significantly higher in comparison with the surfaces. Dissociation of water was found to be highly favorable on the surfaces. The activation barrier for the dissociation of a single water molecule was dependent on the termination of the surface. The electronic properties of and the adsorption behavior of an NO molecule on the (100) surface were studied using DFT+U.124 The authors suggest that the ground state of bulk has an inverse spinel structure and is a magnetic semiconductor. The NO molecule prefers to adsorb on the top site of the Fe atom on the (100) surface, forming an N-Fe bond. DFT+U calculations of the adsorption behavior of Ni and Ti on the surface have been carried out by Bliem et al.125 For both atoms, an incorporation in an octahedral Fe site of the force-relaxed surface is energetically favorable. Boron adsorption on an surface was studied by GGA calculations in 2015.126 It was shown that B adsorption induces half-metallicity at the surface. The adsorption of group IV atoms (C, Si, Ge, Sn) on the surface has been investigated using GGA.127 The results show that all these atoms prefer to bind on the surface oxygen atom, which has no tetrahedral Fe neighbor. The adsorption structures and energies of a single Au atom on six different terminations of the surface were computed using GGA+U.128 It was found that the Au-atom adsorption energy decreases with increasing stability of the surface. Furthermore, the results indicate that the Au atom is reduced and has a negative charge on the iron-terminated surfaces, whereas it is oxidized and has a positive charge on the oxygen-terminated surfaces. Van Natter et al.129 investigated with cluster models possible active sites on the (100), (110), and (111) surfaces of . Adsorption energies of oxygen adatoms located on exposed cation sites were calculated on hybrid-DFT (B3LYP) level of theory. The computed energies vary proportionally to the number of oxygen atoms missing from the normal octahedral coordination of the cation adsorption sites. A theoretical investigation of bare and water terminated surfaces was carried out in 2014 using GGA+U.130 It was found that surfaces that have more metal cations exposed are more stable. The most stable surfaces are shown to be along the (111) planes. Water adsorption on the surfaces was found to be an exothermic process. In 2014, a DFT investigation of the surface reported an overpotential of 0.42 V for the OER.131 It was concluded that Fe-doped -NiOOH and could be the phases responsible for the enhanced OER activity of when it is doped with Fe. 4.ConclusionAs many ferrites are composed of metals with known electrocatalytic properties for the OER,41 the development of cheap and varied electrode materials for PECs is a reachable target without the use of precious OER catalysts like or . However, the existing photoelectrochemical performances need to be enhanced for economically viable PEC applications. Though the fundamental semiconductor properties are satisfactorily understood, the mechanism of charge transport and the water oxidation at the surface is not well known yet. The improvement of new synthesis strategies enabling the formation of high crystalline materials at relatively low temperature (e.g., hydrothermal and microwave synthesis) is crucial to obtain high surface area materials. In addition, thin under layers or over layers of and (but also other metal oxides) are expected to enhance the photocurrent either through adjusting band alignments or passivating surface recombination centers as was revealed for other iron-based photoelectrodes.132 The formation of nanostructures with well-defined morphology, shape, and orientation will enhance the photoactivity by providing a high density of surface reaction sites and by reducing charge recombination as the size of nanostructures approaches the width of the space charge layer. Another strategy which can be exploited further is the use of heterojunctions to effectively separate the photogenerated charge carries with properly matched valence and conduction band edges. Furthermore, existing computational methods adequately predict the electronic and magnetic properties of spinel ferrites. The calculated bandgaps for some of them reasonably agree with experimental values, but for others, more refinements are needed. Most importantly, the relative band positions are key properties to determine the oxidation or reduction power, thus developing computational tools to predict the band positions is essential to understand the very scattered experimentally reported band positions of spinel ferrites. AcknowledgmentsThis work is supported by the Deutsche Forschungsgemeinschaft (DFG) under the program SPP 1613 (Wa 1116/28, BR 1768/9-1, BA 1137/22-1) ReferencesN. S. Lewis and D. G. Nocera,
“Powering the planet: chemical challenges in solar energy utilization,”
Proc. Natl. Acad. Sci., 103
(43), 15729
–15735
(2006). http://dx.doi.org/10.1073/pnas.0603395103 Google Scholar
A. Kudo and Y. Miseki,
“Heterogeneous photocatalyst materials for water splitting,”
Chem. Soc. Rev., 38
(1), 253
–278
(2009). http://dx.doi.org/10.1039/B800489G CSRVBR 0306-0012 Google Scholar
K. Maeda and K. Domen,
“Photocatalytic water splitting: recent progress and future challenges,”
J. Phys. Chem. Lett., 1
(18), 2655
–2661
(2010). http://dx.doi.org/10.1021/jz1007966 JPCLCD 1948-7185 Google Scholar
S. Choudhary et al.,
“Nanostructured bilayered thin films in photoelectrochemical water splitting—a review,”
Int. J. Hydrogen Energy, 37
(24), 18713
–18730
(2012). http://dx.doi.org/10.1016/j.ijhydene.2012.10.028 IJHEDX 0360-3199 Google Scholar
M. S. Prevot and K. Sivula,
“Photoelectrochemical tandem cells for solar water splitting,”
J. Phys. Chem. C, 117
(35), 17879
–17893
(2013). http://dx.doi.org/10.1021/jp405291g JPCCCK 1932-7447 Google Scholar
F. E. Osterloh,
“Inorganic nanostructures for photoelectrochemical and photocatalytic water splitting,”
Chem. Soc. Rev., 42
(6), 2294
–2320
(2013). http://dx.doi.org/10.1039/C2CS35266D CSRVBR 0306-0012 Google Scholar
A. A. Ismail and D. W. Bahnemann,
“Photochemical splitting of water for hydrogen production by photocatalysis: a review,”
Sol. Energy Mater. Sol. Cells, 128 85
–101
(2014). http://dx.doi.org/10.1016/j.solmat.2014.04.037 SEMCEQ 0927-0248 Google Scholar
R. Marschall,
“Semiconductor composites: strategies for enhancing charge carrier separation to improve photocatalytic activity,”
Adv. Funct. Mater., 24
(17), 2421
–2440
(2014). http://dx.doi.org/10.1002/adfm.201303214 AFMDC6 1616-301X Google Scholar
T. Hisatomi, J. Kubota and K. Domen,
“Recent advances in semiconductors for photocatalytic and photoelectrochemical water splitting,”
Chem. Soc. Rev., 43
(22), 7520
–7535
(2014). http://dx.doi.org/10.1039/C3CS60378D CSRVBR 0306-0012 Google Scholar
S. J. A. Moniz et al.,
“Visible-light driven heterojunction photocatalysts for water splitting—a critical review,”
Energy Environ. Sci., 8
(3), 731
–759
(2015). http://dx.doi.org/10.1039/C4EE03271C EESNBY 1754-5692 Google Scholar
M. Sugimoto,
“The past, present, and future of ferrites,”
J. Am. Ceram. Soc., 82
(2), 269
–280
(1999). http://dx.doi.org/10.1111/j.1551-2916.1999.tb20058.x JACTAW 0002-7820 Google Scholar
D. S. Mathew and R. S. Juang,
“An overview of the structure and magnetism of spinel ferrite nanoparticles and their synthesis in microemulsions,”
Chem. Eng. J., 129
(1–3), 51
–65
(2007). http://dx.doi.org/10.1016/j.cej.2006.11.001 Google Scholar
C. Z. Yuan et al.,
“Mixed transition-metal oxides: design, synthesis, and energy-related applications,”
Angew. Chem. Int. Ed., 53
(6), 1488
–1504
(2014). http://dx.doi.org/10.1002/anie.201303971 Google Scholar
M. S. Park et al.,
“Porous nanoarchitectures of spinel-type transition metal oxides for electrochemical energy storage systems,”
Phys. Chem. Chem. Phys., 17
(46), 30963
–30977
(2015). http://dx.doi.org/10.1039/C5CP05936D Google Scholar
E. Casbeer, V. K. Sharma and X. Z. Li,
“Synthesis and photocatalytic activity of ferrites under visible light: a review,”
Sep. Purif. Technol., 87 1
–14
(2012). http://dx.doi.org/10.1016/j.seppur.2011.11.034 Google Scholar
R. Dillert et al.,
“Research update: photoelectrochemical water splitting and photocatalytic hydrogen production using ferrites () under visible light irradiation,”
APL Mater., 3
(10), 104001
(2015). http://dx.doi.org/10.1063/1.4931763 Google Scholar
B. A. Pinaud et al.,
“Technical and economic feasibility of centralized facilities for solar hydrogen production via photocatalysis and photoelectrochemistry,”
Energy Environ. Sci., 6
(7), 1983
–2002
(2013). http://dx.doi.org/10.1039/c3ee40831k EESNBY 1754-5692 Google Scholar
M. Woodhouse and B. A. Parkinson,
“Combinatorial approaches for the identification and optimization of oxide semiconductors for efficient solar photoelectrolysis,”
Chem. Soc. Rev., 38
(1), 197
–210
(2009). http://dx.doi.org/10.1039/B719545C CSRVBR 0306-0012 Google Scholar
M. F. Weber and M. J. Dignam,
“Efficiency of splitting water with semiconducting photoelectrodes,”
J. Electrochem. Soc., 131
(6), 1258
–1265
(1984). http://dx.doi.org/10.1149/1.2115797 JESOAN 0013-4651 Google Scholar
A. B. Murphy et al.,
“Efficiency of solar water splitting using semiconductor electrodes,”
Int. J. Hydrogen Energy, 31
(14), 1999
–2017
(2006). http://dx.doi.org/10.1016/j.ijhydene.2006.01.014 IJHEDX 0360-3199 Google Scholar
J. R. Bolton, S. J. Strickler and J. S. Connolly,
“Limiting and realizable efficiencies of solar photolysis of water,”
Nature, 316
(6028), 495
–500
(1985). http://dx.doi.org/10.1038/316495a0 Google Scholar
M. G. Walter et al.,
“Solar water splitting cells,”
Chem. Rev., 110
(11), 6446
–6473
(2010). http://dx.doi.org/10.1021/cr1002326 CHREAY 0009-2665 Google Scholar
M. Gratzel,
“Photoelectrochemical cells,”
Nature, 414
(6861), 338
–344
(2001). http://dx.doi.org/10.1038/35104607 Google Scholar
A. L. Tiano et al.,
“Correlating size and composition-dependent effects with magnetic, mossbauer, and pair distribution function measurements in a family of catalytically active ferrite nanoparticles,”
Chem. Mater., 27
(10), 3572
–3592
(2015). http://dx.doi.org/10.1021/acs.chemmater.5b00767 CMATEX 0897-4756 Google Scholar
A. Goldman, Modern Ferrite Technology, Springer, Pittsburgh, Pennsylvania
(2006). Google Scholar
D. Levy et al.,
“Equation of state, structural behaviour and phase diagram of synthetic , as a function of pressure and temperature,”
Phys. Chem. Miner., 31
(2), 122
–129
(2004). http://dx.doi.org/10.1007/s00269-004-0380-4 PCMIDU 0342-1791 Google Scholar
R. A. Candeia et al.,
“Synthesis and characterization of spinel pigment obtained by the polymeric precursor method,”
Mater. Lett., 58
(5), 569
–572
(2004). http://dx.doi.org/10.1016/S0167-577X(03)00563-9 MLETDJ 0167-577X Google Scholar
R. A. Candeia et al.,
“Monoferrite applied as ceramic pigment,”
Ceram. Int., 33
(4), 521
–525
(2007). http://dx.doi.org/10.1016/j.ceramint.2005.10.018 Google Scholar
N. Helaili et al.,
“Synthesis and physical properties of the () solid solution,”
Mater. Chem. Phys., 148
(3), 734
–743
(2014). http://dx.doi.org/10.1016/j.matchemphys.2014.08.042 MCHPDR 0254-0584 Google Scholar
W. T. Thompson et al., Uhlig’s Corrosion Handbook, 3rd ed.John Wiley & Sons, Inc., Hoboken, New Jersey
(2011). Google Scholar
Y. Hemmi et al.,
“Electrochemical considerations regarding general corrosion of materials in a bwr primary circuit,”
J. Nucl. Sci. Technol., 31
(11), 1202
–1213
(1994). http://dx.doi.org/10.1080/18811248.1994.9735277 JNSTAX 0022-3131 Google Scholar
Y.-F. Xu et al.,
“In situ formation of zinc ferrite modified Al-doped ZnO nanowire arrays for solar water splitting,”
J. Mater. Chem. A, 4
(14), 5124
–5129
(2016). http://dx.doi.org/10.1039/C5TA10563C Google Scholar
A. A. Tahir and K. G. U. Wijayantha,
“Photoelectrochemical water splitting at nanostructured electrodes,”
J. Photochem. Photobiol. A, 216
(2–3), 119
–125
(2010). http://dx.doi.org/10.1016/j.jphotochem.2010.07.032 JPPCEJ 1010-6030 Google Scholar
J. H. Kim et al.,
“Defective nanorods with oxygen vacancy for photoelectrochemical water splitting,”
Nanoscale, 7
(45), 19144
–19151
(2015). http://dx.doi.org/10.1039/C5NR05812K NANOHL 2040-3364 Google Scholar
A. G. Hufnagel et al.,
“Zinc ferrite photoanode nanomorphologies with favorable kinetics for water-splitting,”
Adv. Funct. Mater., 26
(25), 4435
–4443
(2016). http://dx.doi.org/10.1002/adfm.v26.25 AFMDC6 1616-301X Google Scholar
J. Y. Cao et al.,
“Fabrication of p-type nanofilms for photoelectrochemical hydrogen generation,”
Electrochem. Commun., 13
(3), 275
–278
(2011). http://dx.doi.org/10.1016/j.elecom.2011.01.002 ECCMF9 1388-2481 Google Scholar
S. Ida et al.,
“Photoelectrochemical hydrogen production from water using p-type and n-type oxide semiconductor electrodes,”
Electrochim. Acta, 82 397
–401
(2012). http://dx.doi.org/10.1016/j.electacta.2012.03.174 ELCAAV 0013-4686 Google Scholar
S. Ida et al.,
“Preparation of p-Type photocathodes for producing hydrogen from water,”
J. Am. Chem. Soc., 132
(49), 17343
–17345
(2010). http://dx.doi.org/10.1021/ja106930f JACSAT 0002-7863 Google Scholar
H. Yang et al.,
“Electrochemical synthesis of porous nanosheets for visible light driven photoelectrochemical applications,”
New J. Chem., 37
(10), 2965
–2968
(2013). http://dx.doi.org/10.1039/c3nj00627a NJCHE5 1144-0546 Google Scholar
G. Rekhila, Y. Bessekhouad and M. Trari,
“Visible light hydrogen production on the novel ferrite ,”
Int. J. Hydrogen Energy, 38
(15), 6335
–6343
(2013). http://dx.doi.org/10.1016/j.ijhydene.2013.03.087 IJHEDX 0360-3199 Google Scholar
Y. Matsumoto et al.,
“Photoelectrochemical properties of the Zn-Ti-Fe spinel oxides,”
J. Electrochem. Soc., 133
(4), 711
–716
(1986). http://dx.doi.org/10.1149/1.2108660 JESOAN 0013-4651 Google Scholar
Z. Simsa et al.,
“Optical and magneto-optical properties of magnetite and manganese ferrites,”
J. Magn. Magn. Mater., 15–18 775
–776
(1980). http://dx.doi.org/10.1016/0304-8853(80)90757-X JMMMDC 0304-8853 Google Scholar
S. Balaji et al.,
“Combustion synthesis and characterization of substituted nanocrystalline ,”
Mater. Sci. Eng. B, 119
(2), 119
–124
(2005). http://dx.doi.org/10.1016/j.mseb.2005.01.021 Google Scholar
K. N. Harish et al.,
“Synthesis, enhanced optical and photocatalytic study of Cd-Zn ferrites under sunlight,”
Catal. Sci. Technol., 2
(5), 1033
–1039
(2012). http://dx.doi.org/10.1039/c2cy00503d Google Scholar
M. D. Archer, G. C. Morris and G. K. Yim,
“Electrochemical approaches to solar-energy conversion—a brief overview and preliminary-results obtained with n-type cobalt ferrite,”
J. Electroanal. Chem., 118 89
–100
(1981). http://dx.doi.org/10.1016/S0022-0728(81)80534-7 JECHES 0022-0728 Google Scholar
M. S. Antonious et al.,
“Photoelectrochemical characteristics of p-type and n-type polycrystalline Ni-ferrite electrodes in aqueous-solutions,”
Mater. Res. Bull., 21
(12), 1515
–1523
(1986). http://dx.doi.org/10.1016/0025-5408(86)90093-0 MRBUAC 0025-5408 Google Scholar
C. G. Ramankutty and S. Sugunan,
“Surface properties and catalytic activity of ferrospinels of nickel, cobalt and copper, prepared by soft chemical methods,”
Appl. Catal. A, 218
(1–2), 39
–51
(2001). http://dx.doi.org/10.1016/S0926-860X(01)00610-X Google Scholar
G. K. Reddy et al.,
“Cr- and Ce-doped ferrite catalysts for the high temperature water-gas shift reaction: TPR and Mossbauer spectroscopic study,”
J. Phys. Chem. C, 115
(4), 920
–930
(2011). http://dx.doi.org/10.1021/jp102959p JPCCCK 1932-7447 Google Scholar
Y. Matsumoto,
“Energy positions of oxide semiconductors and photocatalysis with iron complex oxides,”
J. Solid State Chem., 126
(2), 227
–234
(1996). http://dx.doi.org/10.1006/jssc.1996.0333 JSSCBI 0022-4596 Google Scholar
H. H. Kung et al.,
“Semiconducting oxide anodes in photoassisted electrolysis of water,”
J. Appl. Phys., 48
(6), 2463
–2469
(1977). http://dx.doi.org/10.1063/1.324010 JAPIAU 0021-8979 Google Scholar
L. G. J. Dehaart and G. Blasse,
“Photoelectrochemical properties of ferrites with the spinel structure,”
J. Electrochem. Soc., 132
(12), 2933
–2938
(1985). http://dx.doi.org/10.1149/1.2113696 JESOAN 0013-4651 Google Scholar
F. A. Benko and F. P. Koffyberg,
“The effect of defects on some photoelectrochemical properties of semiconducting ,”
Mater. Res. Bull., 21
(10), 1183
–1188
(1986). http://dx.doi.org/10.1016/0025-5408(86)90045-0 MRBUAC 0025-5408 Google Scholar
H. Zazoua et al.,
“Enhanced photocatalytic hydrogen production under visible light over a material based on magnesium ferrite derived from layered double hydroxides (LDHs),”
Int.J. Energy Res., 38
(15), 2010
–2018
(2014). http://dx.doi.org/10.1002/er.v38.15 IJERDN 0363-907X Google Scholar
M. Buchler et al.,
“Comparison of the semiconductive properties of sputter-deposited iron oxides with the passive film on iron,”
J. Electrochem. Soc., 145
(2), 378
–385
(1998). http://dx.doi.org/10.1149/1.1838272 JESOAN 0013-4651 Google Scholar
Y. Matsumoto et al.,
“New photocathode materials for hydrogen evolution— and ,”
J. Phys. Chem., 91
(3), 577
–581
(1987). http://dx.doi.org/10.1021/j100287a018 JPCHAX 0022-3654 Google Scholar
Y. Matsumoto, K. Sugiyama and E. I. Sato,
“Improvement of photocathode by doping with Na and Mg,”
J. Solid State Chem., 74
(1), 117
–125
(1988). http://dx.doi.org/10.1016/0022-4596(88)90337-4 JSSCBI 0022-4596 Google Scholar
S. Ida et al.,
“Photoelectrochemical hydrogen production from water using p-type and n-Type ZnO,”
Electrochemistry, 79
(10), 797
–800
(2011). http://dx.doi.org/10.5796/electrochemistry.79.797 ECHMBU 0305-9979 Google Scholar
J. Y. Cao et al.,
“Photoelectrochemical properties of nanomultiple pn junction photoelectrodes,”
Langmuir, 29
(9), 3116
–3124
(2013). http://dx.doi.org/10.1021/la304377z LANGD5 0743-7463 Google Scholar
K. Sekizawa et al.,
“Structural improvement of by metal doping toward enhanced cathodic photocurrent,”
ACS Appl. Mater. Interfaces, 6
(14), 10969
–10973
(2014). http://dx.doi.org/10.1021/am502500y AAMICK 1944-8244 Google Scholar
B. T. Chang et al.,
“Photoelectrochemical study of a spinel-type titanomagnetite,”
J. Solid State Chem., 72
(2), 201
–208
(1988). http://dx.doi.org/10.1016/0022-4596(88)90023-0 JSSCBI 0022-4596 Google Scholar
A. A. Tahir et al.,
“A new route to control texture of materials: nanostructured photoelectrodes,”
Int. J. Hydrogen Energy, 38
(11), 4315
–4323
(2013). http://dx.doi.org/10.1016/j.ijhydene.2013.01.130 IJHEDX 0360-3199 Google Scholar
K. Dileep et al.,
“Probing optical band gaps at the nanoscale in and epitaxial films by high resolution electron energy loss spectroscopy,”
J. Appl. Phys., 116
(10), 103505
(2014). http://dx.doi.org/10.1063/1.4895059 JAPIAU 0021-8979 Google Scholar
C. Himcinschi et al.,
“Optical and magneto-optical study of nickel and cobalt ferrite epitaxial thin films and submicron structures,”
J. Appl. Phys., 113
(8), 084101
(2013). http://dx.doi.org/10.1063/1.4792749 JAPIAU 0021-8979 Google Scholar
P. Xiong et al.,
“Ternary titania-cobalt ferrite-polyaniline nanocomposite: a magnetically recyclable hybrid for adsorption and photodegradation of dyes under visible light,”
Ind. Eng. Chem. Res., 52
(30), 10105
–10113
(2013). http://dx.doi.org/10.1021/ie400739e Google Scholar
J. H. Kim et al.,
“Awakening solar water-splitting activity of nanorods by hybrid microwave annealing,”
Adv. Energy Mater., 5
(6),
(2015). http://dx.doi.org/10.1002/aenm.201401933 ADEMBC 1614-6840 Google Scholar
K. J. McDonald and K. S. Choi,
“Synthesis and photoelectrochemical properties of composite photoanodes for use in solar water oxidation,”
Chem. Mater., 23
(21), 4863
–4869
(2011). http://dx.doi.org/10.1021/cm202399g CMATEX 0897-4756 Google Scholar
R. Dom et al.,
“Eco-friendly ferrite nanocomposite photoelectrode for improved solar hydrogen generation,”
RSC Adv., 3
(35), 15217
–15224
(2013). http://dx.doi.org/10.1039/c3ra42051e Google Scholar
Y. H. Guo et al.,
“Photoelectrochemical activity of modified alpha- nanorod array films,”
RSC Adv., 4
(70), 36967
–36972
(2014). http://dx.doi.org/10.1039/C4RA05289G Google Scholar
M. G. Ahmed et al.,
“Enhanced photoelectrochemical water oxidation on nanostructured hematite photoanodes via p- heterojunction formation,”
J. Phys. Chem. C, 119
(11), 5864
–5871
(2015). http://dx.doi.org/10.1021/jp512804p JPCCCK 1932-7447 Google Scholar
J. Y. Kim et al.,
“Single-crystalline, wormlike hematite photoanodes for efficient solar water splitting,”
Sci. Rep., 3 2681
(2013). http://dx.doi.org/10.1038/srep02681 Google Scholar
E. S. Kim et al.,
“Fabrication of heterojunction photoanode for photoelectrochemical water oxidation,”
J. Am. Chem. Soc., 135
(14), 5375
–5383
(2013). http://dx.doi.org/10.1021/ja308723w JACSAT 0002-7863 Google Scholar
H. G. Kim et al.,
“Fabrication of bulk heterojunction for enhanced visible light photocatalysis,”
Chem. Commun., 39 5889
–5891
(2009). http://dx.doi.org/10.1039/b911805e Google Scholar
X. L. Zheng et al.,
“ leaves grown on trees enhance photoelectrochemical water splitting,”
Small, 12
(23), 3181
–3188
(2016). http://dx.doi.org/10.1002/smll.v12.23 SMALBC 1613-6810 Google Scholar
E. S. Kim et al.,
“Improved photoelectrochemical activity of heterojunction photoanode by reduced surface recombination in solar water oxidation,”
ACS Appl. Mater. Interfaces, 6
(20), 17762
–17769
(2014). http://dx.doi.org/10.1021/am504283t AAMICK 1944-8244 Google Scholar
M. W. Kanan and D. G. Nocera,
“In situ formation of an oxygen-evolving catalyst in neutral water containing phosphate and ,”
Science, 321
(5892), 1072
–1075
(2008). http://dx.doi.org/10.1126/science.1162018 Google Scholar
M. W. Kanan, Y. Surendranath and D. G. Nocera,
“Cobalt-phosphate oxygen-evolving compound,”
Chem. Soc. Rev., 38
(1), 109
–114
(2009). http://dx.doi.org/10.1039/B802885K CSRVBR 0306-0012 Google Scholar
Y. Surendranath, M. W. Kanan and D. G. Nocera,
“Mechanistic studies of the oxygen evolution reaction by a cobalt-phosphate catalyst at neutral pH,”
J. Am. Chem. Soc., 132
(46), 16501
–16509
(2010). http://dx.doi.org/10.1021/ja106102b JACSAT 0002-7863 Google Scholar
G. M. Carroll, D. K. Zhong and D. R. Gamelin,
“Mechanistic insights into solar water oxidation by cobalt-phosphate-modified alpha- photoanodes,”
Energy Environ. Sci., 8
(2), 577
–584
(2015). http://dx.doi.org/10.1039/C4EE02869D EESNBY 1754-5692 Google Scholar
A. Gruneis et al.,
“Ionization potentials of solids: the importance of vertex corrections,”
Phys. Rev. Lett., 112
(9), 096401
(2014). http://dx.doi.org/10.1103/PhysRevLett.112.096401 PRLTAO 0031-9007 Google Scholar
M. Marsman et al.,
“Hybrid functionals applied to extended systems,”
J. Phys: Condens. Matter, 20
(6), 064201
(2008). http://dx.doi.org/10.1088/0953-8984/20/6/064201 Google Scholar
S. L. Dudarev et al.,
“Electron-energy-loss spectra and the structural stability of nickel oxide: An LSDA+U study,”
Phys. Rev. B, 57
(3), 1505
–1509
(1998). http://dx.doi.org/10.1103/PhysRevB.57.1505 Google Scholar
D. Odkhuu et al.,
“A first-principles study of magnetostrictions of and ,”
J. Appl. Phys., 115
(17), 17A916
(2014). http://dx.doi.org/10.1063/1.4863811 JAPIAU 0021-8979 Google Scholar
K. Obata et al.,
“Electronic structure of with antiferromagnetic spin ordering,”
J. Ceram. Soc. Jpn., 121
(1417), 766
–769
(2013). http://dx.doi.org/10.2109/jcersj2.121.766 JCSJEW 0914-5400 Google Scholar
D. H. Ji et al.,
“Quantum mechanical method for estimating ionicity of spinel ferrites,”
J. Magn. Magn. Mater., 326 197
–200
(2013). http://dx.doi.org/10.1016/j.jmmm.2012.09.016 JMMMDC 0304-8853 Google Scholar
Q. C. Sun et al.,
“Optical band gap hierarchy in a magnetic oxide: electronic structure of ,”
Phys. Rev. B, 86
(20), 205106
(2012). http://dx.doi.org/10.1103/PhysRevB.86.205106 Google Scholar
R. Dom et al.,
“Synthesis of solar active nanocrystalline ferrite, (M: Ca, Zn, Mg) photocatalyst by microwave irradiation,”
Solid State Commun., 151
(6), 470
–473
(2011). http://dx.doi.org/10.1016/j.ssc.2010.12.034 SSCOA4 0038-1098 Google Scholar
S. Soliman et al.,
“Electronic structure calculations for ,”
Phys. Rev. B, 83
(8), 085205
(2011). http://dx.doi.org/10.1103/PhysRevB.83.085205 Google Scholar
M. Feng et al.,
“Ab initio study on copper ferrite,”
J. Appl. Phys., 107
(9), 09A521
(2010). http://dx.doi.org/10.1063/1.3338905 JAPIAU 0021-8979 Google Scholar
D. M. Wells et al.,
“Local electronic and magnetic structure of mixed ferrite multilayer materials,”
Phys. Rev. B, 81
(17), 174422
(2010). http://dx.doi.org/10.1103/PhysRevB.81.174422 Google Scholar
C. Cheng and C.-S. Liu,
“Effects of cation distribution in and : ab initio studies,”
J. Phys.: Conf. Ser, 145 012028
(2009). http://dx.doi.org/10.1088/1742-6596/145/1/012028 JPCSDZ 1742-6588 Google Scholar
X. Zuo et al.,
“A computational study of nickel ferrite,”
J. Magn. Magn. Mater., 303
(2), E432
–E435
(2006). http://dx.doi.org/10.1016/j.jmmm.2006.01.102 JMMMDC 0304-8853 Google Scholar
Z. Szotek et al.,
“Electronic structures of normal and inverse spinel ferrites from first principles,”
Phys. Rev. B, 74
(17), 174431
(2006). http://dx.doi.org/10.1103/PhysRevB.74.174431 Google Scholar
V. N. Antonov, B. N. Harmon and A. N. Yaresko,
“Electronic structure and x-ray magnetic circular dichroism in and Mn-, Co-, or Ni-substituted ,”
Phys. Rev. B, 67
(2), 024417
(2003). http://dx.doi.org/10.1103/PhysRevB.67.024417 Google Scholar
X. Zuo and C. Vittoria,
“Calculation of exchange integrals and electronic structure for manganese ferrite,”
Phys. Rev. B, 66
(18), 184420
(2002). http://dx.doi.org/10.1103/PhysRevB.66.184420 Google Scholar
D. J. Singh, M. Gupta and R. Gupta,
“First-principles investigation of ,”
Phys. Rev. B, 65
(6), 064432
(2002). http://dx.doi.org/10.1103/PhysRevB.65.064432 Google Scholar
N. Y. Safontseva and I. Y. Nikiforov,
“On the shape of iron K absorption edges for monoferrites with a Me(Mg, Mn, Ni, Zn) spinel structure,”
Phys. Solid State, 43
(1), 61
–64
(2001). http://dx.doi.org/10.1134/1.1340188 PSOSED 1063-7834 Google Scholar
W. F. Pong et al.,
“Oxygen 1s x-ray-absorption near-edge structure of Zn-Ni ferrites: a comparison with the theoretical calculations,”
Phys. Rev. B, 54
(23), 16641
–16645
(1996). http://dx.doi.org/10.1103/PhysRevB.54.16641 Google Scholar
W. C. Mackrodt and E. A. Simson,
“Cation valence charge states of : an ab initio Hartree-Fock study,”
J. Chem. Soc. Faraday Trans., 92
(12), 2043
–2047
(1996). http://dx.doi.org/10.1039/FT9969202043 Google Scholar
M. Penicaud et al.,
“Calculated electronic band-structure and magnetic-moments of ferrites,”
J. Magn. Magn. Mater., 103
(1–2), 212
–220
(1992). http://dx.doi.org/10.1016/0304-8853(92)90255-M JMMMDC 0304-8853 Google Scholar
K. Jordan et al.,
“Scanning tunneling spectroscopy study of the electronic structure of Fe3O4 surfaces,”
Phys. Rev. B, 74
(8), 085416
(2006). http://dx.doi.org/10.1103/PhysRevB.74.085416 Google Scholar
D. A. Andersson and C. R. Stanek,
“Mixing and non-stoichiometry in Fe-Ni-Cr-Zn-O spinel compounds: density functional theory calculations,”
Phys. Chem. Chem. Phys., 15
(37), 15550
–15564
(2013). http://dx.doi.org/10.1039/c3cp50312g PPCPFQ 1463-9076 Google Scholar
H. T. Jeng, G. Y. Guo and D. J. Huang,
“Charge-orbital ordering and Verwey transition in magnetite,”
Phys. Rev. Lett., 93
(15), 156403
(2004). http://dx.doi.org/10.1103/PhysRevLett.93.156403 PRLTAO 0031-9007 Google Scholar
I. Leonov et al.,
“Electronic structure of charge-ordered from calculated optical, magneto-optical Kerr effect, and OK-edge x-ray absorption spectra,”
Phys. Rev. B, 74
(16), 165117
(2006). http://dx.doi.org/10.1103/PhysRevB.74.165117 Google Scholar
A. D. Rowan, C. H. Patterson and L. V. Gasparov,
“Hybrid density functional theory applied to magnetite: crystal structure, charge order, and phonons,”
Phys. Rev. B, 79
(20), 205103
(2009). http://dx.doi.org/10.1103/PhysRevB.79.205103 Google Scholar
G. K. H. Madsen and P. Novak,
“Charge order in magnetite. An LDA+U study,”
Europhys. Lett., 69
(5), 777
–783
(2005). http://dx.doi.org/10.1209/epl/i2004-10416-x EULEEJ 0295-5075 Google Scholar
Z. Szotek et al.,
“Ab initio study of charge order in ,”
Phys. Rev. B, 68
(5), 054415
(2003). http://dx.doi.org/10.1103/PhysRevB.68.054415 Google Scholar
H. G. Kim et al.,
“Photocatalytic nanodiodes for visible-light photocatalysis,”
Angew. Chem. Int. Ed., 44
(29), 4585
–4589
(2005). http://dx.doi.org/10.1002/(ISSN)1521-3773 Google Scholar
P. H. Borse et al.,
“Synthesis of barium ferrite for visible light photocatalysis applications,”
J. Korean Phys. Soc., 58
(6), 1672
–1676
(2011). http://dx.doi.org/10.3938/jkps.58.1672 KPSJAS 0374-4884 Google Scholar
C. J. O’Brien, Z. Rak and D. W. Brenner,
“Free energies of (Co, Fe, Ni, Zn) spinels and oxides in water at high temperatures and pressure from density functional theory: results for stoichiometric NiO and surfaces,”
J. Phys.: Condens. Matter, 25
(44), 445008
(2013). http://dx.doi.org/10.1088/0953-8984/25/44/445008 Google Scholar
D. Fritsch and C. Ederer,
“Epitaxial strain effects in the spinel ferrites and from first principles,”
Phys. Rev. B, 82
(10), 104117
(2010). http://dx.doi.org/10.1103/PhysRevB.82.104117 Google Scholar
Z. Rak, C. J. O’Brien and D. W. Brenner,
“First-principles investigation of boron defects in nickel ferrite spinel,”
J. Nucl. Mater., 452
(1–3), 446
–452
(2014). http://dx.doi.org/10.1016/j.jnucmat.2014.05.031 JNUMAM 0022-3115 Google Scholar
A. Kezzim et al.,
“Visible light induced hydrogen on the novel hetero-system ,”
Energy Convers. Manage., 52
(8–9), 2800
–2806
(2011). http://dx.doi.org/10.1016/j.enconman.2011.02.014 ECMADL 0196-8904 Google Scholar
P. H. Borse et al.,
“Photocatalytic hydrogen generation from water-methanol mixtures using nanocrystalline under visible light irradiation,”
J. Korean Phys. Soc., 55
(4), 1472
–1477
(2009). http://dx.doi.org/10.3938/jkps.55.1472 KPSJAS 0374-4884 Google Scholar
S. Boumaza et al.,
“Visible light induced hydrogen evolution on new hetero-system ,”
Appl. Energy, 87
(7), 2230
–2236
(2010). http://dx.doi.org/10.1016/j.apenergy.2009.12.016 Google Scholar
R. Dom et al.,
“Synthesis of a hydrogen producing nanocrystalline visible light photocatalyst using a rapid microwave irradiation method,”
RSC Adv., 2
(33), 12782
–12791
(2012). http://dx.doi.org/10.1039/c2ra21910g Google Scholar
J. H. Yao et al.,
“Density functional theory investigations on the structure and electronic properties of normal spinel ,”
Integr. Ferroelectr., 145
(1), 17
–23
(2013). http://dx.doi.org/10.1080/10584587.2013.788310 IFEREU 1058-4587 Google Scholar
A. Elfalaky and S. Soliman,
“Theoretical investigation of ,”
J. Alloys Compd., 580 401
–406
(2013). http://dx.doi.org/10.1016/j.jallcom.2013.05.197 JALCEU 0925-8388 Google Scholar
M. Padervand et al.,
“An experimental and theoretical study on the structure and photoactivity of (, Fe, Ni, Co, and Zn) structures,”
Russ. J. Phys. Chem. A, 88
(13), 2451
–2461
(2014). http://dx.doi.org/10.1134/S0036024414130184 Google Scholar
H. M. Widatallah et al.,
“Atomistic simulation and ab initio study of the defect structure of spinel-related ,”
Mater. Res. Bull., 47
(12), 3995
–4000
(2012). http://dx.doi.org/10.1016/j.materresbull.2012.08.048 MRBUAC 0025-5408 Google Scholar
Y. H. Hou et al.,
“Structural, electronic and magnetic properties of partially inverse spinel CoFe2O4: a first-principles study,”
J. Phys. D: Appl. Phys., 43
(44), 445003
(2010). http://dx.doi.org/10.1088/0022-3727/43/44/445003 JPAPBE 0022-3727 Google Scholar
D. Santos-Carballal et al.,
“First-principles study of the inversion thermodynamics and electronic structure of (thio) spinels (, Mn, Co, Ni; , S),”
Phys. Rev. B, 91
(19), 195106
(2015). http://dx.doi.org/10.1103/PhysRevB.91.195106 Google Scholar
X. Shi et al.,
“Structure of the surface in contact with gaseous and water vapor,”
Surf. Sci., 640 73
–79
(2015). http://dx.doi.org/10.1016/j.susc.2015.03.012 SUSCAS 0039-6028 Google Scholar
P. V. Kumar et al.,
“High surface reactivity and water adsorption on (111) surfaces,”
J. Phys. Chem. C, 117
(11), 5678
–5683
(2013). http://dx.doi.org/10.1021/jp309434a JPCCCK 1932-7447 Google Scholar
Z. Jiang et al.,
“Adsorption of NO molecule on spinel-type surface: a first-principles study,”
J. Phys. Chem. C, 115
(26), 13035
–13040
(2011). http://dx.doi.org/10.1021/jp203492j JPCCCK 1932-7447 Google Scholar
R. Bliem et al.,
“Adsorption and incorporation of transition metals at the magnetite surface,”
Phys. Rev. B, 92
(7), 075440
(2015). http://dx.doi.org/10.1103/PhysRevB.92.075440 Google Scholar
X. Sun, A. Pratt and Y. Yamauchi,
“Half-metallicity induced by boron adsorption on an surface,”
Phys. Chem. Chem. Phys., 17
(23), 15386
–15391
(2015). http://dx.doi.org/10.1039/C5CP02466H PPCPFQ 1463-9076 Google Scholar
X. Sun et al.,
“Significant variation of surface spin polarization through group IV atom (C, Si, Ge, Sn) adsorption on (100),”
Phys. Chem. Chem. Phys., 16
(1), 95
–102
(2014). http://dx.doi.org/10.1039/C3CP53272K PPCPFQ 1463-9076 Google Scholar
X. H. Yu et al.,
“Single gold atom adsorption on the surface,”
J. Phys. Chem. C, 116
(19), 10632
–10638
(2012). http://dx.doi.org/10.1021/jp301313u Google Scholar
R. M. Van Natter, J. S. Coleman and C. R. F. Lund,
“DFT models for active sites on high temperature water-gas shift catalysts,”
J. Mol. Catal. A: Chem., 292
(1–2), 76
–82
(2008). http://dx.doi.org/10.1016/j.molcata.2008.07.015 Google Scholar
C. J. O’Brien, Z. Rak and D. W. Brenner,
“Calculated stability and structure of nickel ferrite crystal surfaces in hydrothermal environments,”
J. Phys. Chem. C, 118
(10), 5414
–5423
(2014). http://dx.doi.org/10.1021/jp5002308 JPCCCK 1932-7447 Google Scholar
Y. F. Li and A. Selloni,
“Mechanism and activity of water oxidation on selected surfaces of pure and Fe-doped ,”
ACS Catal., 4
(4), 1148
–1153
(2014). http://dx.doi.org/10.1021/cs401245q Google Scholar
L. Steier et al.,
“Understanding the role of underlayers and overlayers in thin film hematite photoanodes,”
Adv. Funct. Mater., 24
(48), 7681
–7688
(2014). http://dx.doi.org/10.1002/adfm.v24.48 AFMDC6 1616-301X Google Scholar
BiographyDereje H. Taffa is a postdoctoral research associate in the Institute of Chemistry at the Carl von Ossietzky University Oldenburg. He studied chemistry at Addis Ababa University in Ethiopia and received his BSc degree in chemistry and MSc degree in physical chemistry. He earned his PhD in chemistry from Osnabrueck University. His research interest includes preparation and characterization of oxide based semiconductors thin films for energy storage and conversion applications. Ralf Dillert is working since 2006 as a chemist at the Institute of Technical Chemistry of the Leibniz University Hannover. His actual research interests comprise photocatalytic water treatment, photocatalytic removal of air pollutants, self-cleaning surfaces, and the interfacial electron transfer at the electrolyte/semiconductor interface. Anna C. Ulpe is a PhD student of chemistry in the Mulliken Center for Theoretical Chemistry at the University of Bonn. Her current field of research is the computational investigation of optical properties of semiconductors. Katharina C. L. Bauerfeind studied chemistry and is working towards her PhD in the Mulliken Center for Theoretical Chemistry at the University of Bonn. Currently she is investigating the photocatalytic properties of binary and ternary transition metal oxides with computational means. Thomas Bredow studied chemistry and received his MSc degree and PhD in chemistry from the Leibniz University of Hannover. He is currently a professor of theoretical chemistry in the Mulliken Center for Theoretical Chemistry at the University of Bonn. His research interest includes quantum-chemical modeling of solids and surfaces, defect formation and migration, heterogeneous catalysis, and semiempirical methods. Detlef W. Bahnemann is a supernumerary professor and head of Photocatalysis and Nanotechnology Research Unit at the Institute of Technical Chemistry, Leibniz University of Hannover and the director of Megagrant Laboratory “Photoactive Nanocomposite Materials” at St. Petersburg State University in Russia. He received his MSc degree and PhD in chemistry from the Technical University Berlin. His research focuses on photocatalysis and ultrafast photocatalytic processes, solar water splitting and photoelectrochemical solar cells. Michael Wark is a professor of technical chemistry and the head of the research group of photocatalysis and sustainable feedstock utilization in the Institute of Chemistry at Carl von Ossietzky University Oldenburg. He studied chemistry and obtained his MSc degree and his PhD in chemistry from the University of Bremen. His research interest includes materials for renewable energy applications, micro-and mesoporous structures, ordered mesoporous thin films and inorganic-organic hybrid structures. |



