
|
|
1.IntroductionThis article describes the design rationale for oligomers and polymers with large permanent electric dipoles, i.e., molecular electrets, composed of anthranilamide (Aa) residues (Fig. 1).1–3 A comparison with biopolymers illustrates the basis for referring to the Aa molecular electrets as bioinspired. For improved charge-transfer (CT) properties, the Aa electrets incorporate features from different biomolecular structures to form non-native systems beneficial for electronic and photonic materials for energy applications. Fig. 1Bioinspired molecular electrets composed of anthranilamide (Aa) residues: (a) a segment of a molecular electret—; (b) origin of the Aa dipole from ordered orientation of amide bonds and from the polarization upon hydrogen bonding. 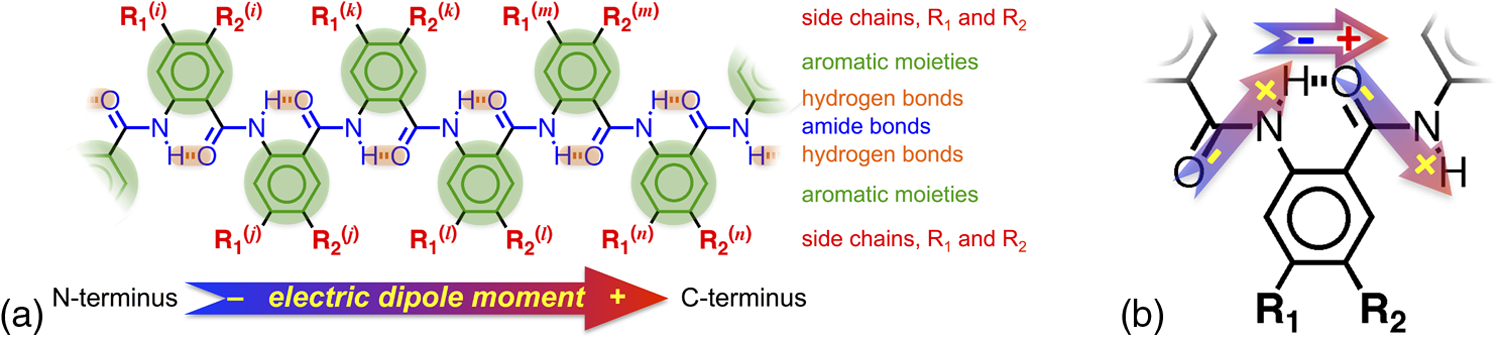 Understanding and controlling CT at a nanometer scale is paramount for solar-energy harvesting, conversion, and storage.4–8 Local electric fields, originating from molecular dipoles, can strongly affect the behavior of charge carriers. Molecular-scale engineering of field profiles along CT pathways provides an incomparable means for controlling the direction and the efficiency of electron and hole transduction. Therefore, molecular electrets can play a central role in the bottom-up designs of hybrid electronic materials and energy-conversion devices. Oligomers and polymers of aromatic -amino acids, i.e., Aa residues, have two uniquely combined features: (1) ordered amide and hydrogen bonds resulting in electric dipoles of about 3 D/residue, oriented from the N- to the C-termini (Fig. 1)9 and (2) extended -conjugation along the macromolecular backbone that can prove essential for mediating long-range CT.10 Our computational and experimental studies demonstrated that Aa conjugates possess permanent electric dipoles, i.e., they are indeed molecular electrets.2,9 Comparing the extent of deshielding (i.e., relative acidity) of the amide protons as determined from nuclear magnetic resonance (NMR) studies provided experimental confirmation that the total molecular dipoles are oriented form the N- to the C-termini of the Aa conjugates.2 We have demonstrated that an Aa residue can act as a molecular rectifier (or a diode), showing preference of electron transfer (ET) toward the C-terminus.1 This emphasis on rectification of CT, rather than charge transport, allows for assessing the effects induced by small organic residues or short oligomers without the interference from the contacts with conducting and semiconducting substrates.11,12 Interfacial charge transport and CT “hold” the key for improving energy-conversion efficiencies of materials and devices.4,13,14 Molecular-level understanding of charge-transfer rectification, implemented with a rational control of surfaces and interfaces,5,15–17 provides the venues for such efficiency improvements. The magnitude of CT rectification for a single Aa residue is comparable with the rectification exerted by peptide helices comprising more than 10 amino acid residues.1 The molecular dipoles have a dominating contribution toward this rectification effect. The asymmetry in electron-density distribution, induced by the positions of the substituents, and the molecular dynamics also contribute to the CT processes. The latter affects the photoinduced charge separation kinetics, while the former affects the consequent charge recombination.1 To truly benefit from the promising CT properties of Aa, it is essential to achieve structural diversity of these electret conjugates, leading to wide variety of electronic functionalities. Extending principles from proteomics to the design of widely diverse molecular electrets places a demand on developing sets of their building blocks, i.e., the non-native Aa residues, with different electronic properties. Herein, we describe the rationale behind bringing structural features from different classes of biomacromolecules (i.e., proteins and nucleic acids) to design Aa molecular electrets in the search of improved CT properties that are not inherent for living systems. We focus on a set of Aa residues that are chemically modified with electron-donating substituents (at and , Fig. 1) for lowering the ionization energies and improving hole-transfer capabilities. Replacing the hydrogen at with three types of substituents with different electron-donating propensity causes negative shifts in the Aa reduction potential, amounting to about 0.1, 0.3, and 1 V. Moving an electron-donating group (EDG) from to results in a positive shift in the reduction potentials of about 0.1 to 0.3 V, which is indicative of an increase in the ionization energy. Discussion of their rectification and other CT properties illustrates the potential impact that the diversity in Aa molecular electrets can have on electronic materials and energy conversion. 2.CT Molecular Electrets: Practical Ideas from Structural BiologyElectrets are dielectrics with permanent electric polarization. The polarization can originate from volume-embedded or surface-trapped charges (i.e., real-charge electrets), or from codirectionally ordered electric dipoles (i.e., dipole-polarization or dipolar electrets).18,19 The lack of directionality of electrostatic interactions and ionic bonds governs the dynamics and the complexity in the behavior of real-charge electrets that encompass “space-charge” and “surface-charge electrets.”20,21 Conversely, the defined directionality of covalent bonds (which, indeed, are also electric in nature) provides a key advantage for arranging noncharged molecular dipolar moieties. For molecular dipoles, patterns of covalent bonding keep the displacement of the centers of positive and negative charges more or less permanently fixed. Therefore, dipolar electrets, which are electrostatic analogues of magnets,22 provide an important venue for the design and development of macromolecules and materials for electronic and energy applications. Biology presents some of the best examples of molecular electrets and the utility of their electronic properties. Peptide bonds are aliphatic amides (with permanent dipoles of about 4 D)23 that hold together the protein backbones. In protein -helices, the peptide bonds are codirectionally oriented along the helix axes (Fig. 2), resulting in substantial electrical polarization of these macromolecular secondary conformers,24–26 i.e., protein -helices are biomolecular electrets. Fig. 2Protein -helix, ribbon diagram and ball and stick model with highlighted hydrogen-bond network. 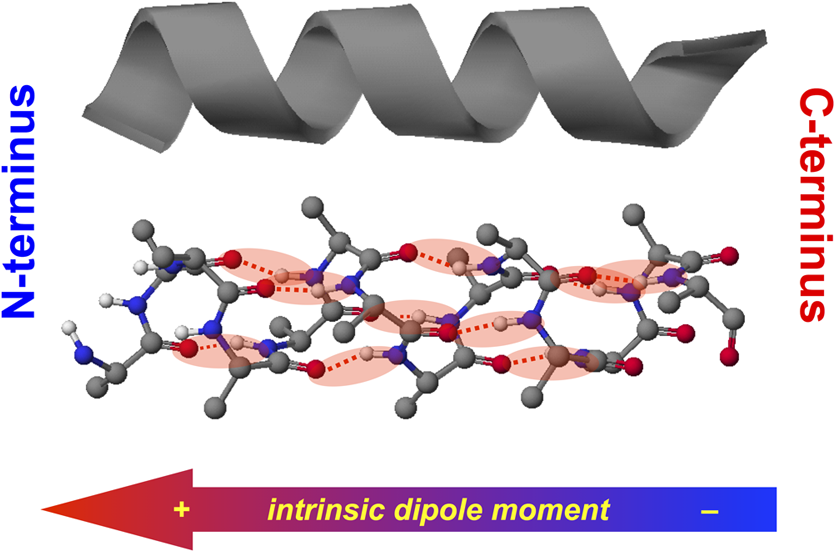 A network of hydrogen bonds, which are also aligned along the helix axes supporting these protein structures (Fig. 2), further enhances the total macromolecular dipoles of protein α-helices. The hydrogen bonding between the carbonyl oxygens and amide hydrogens from peptide bonds on neighboring helix turns causes a collective shift in electron density from the oxygens to the hydrogens, i.e., it causes polarization that is in the direction of the amide dipoles. This effect, however, does not result in negatively charged hydrogens and positively charged oxygens. Rather, the oxygens in the peptide bonds are negatively polarized and upon hydrogen bonding, some of this partial negative charge shifts toward the N–H bond of an amide on the neighboring turn. The ordered hydrogen and peptide bonds in the -helices result in their intrinsic electric dipoles of about 5 D/residue, pointing from the C- to the N-termini (Fig. 2).27 Other protein helices also exhibit dipole-induced polarization. Similar to -helices, alignment of hydrogen and amide bonds in -helices results in intrinsic electric dipoles (4.5 D/residue) pointing from the C- to the N-termini.27 In fact, -helices are tight-wound analogues of -helices: i.e., -helices contain three residues per turn and each hydrogen bond closes a backbone loop comprising 10 atoms. Indeed, protein -helices are helices. Polyproline helices exhibit another interesting set of electronic properties. Proline is a native amino acid with a rigid structure and a secondary -amine. Therefore, polyprolines assume helical conformations without hydrogen bonding, making their intrinsic dipoles smaller than the dipoles of -helices.27 In the two types of polyproline helices, type I and type II, the peptide bonds have different conformations and their dipoles are orientated differently. Type I comprises “cis” amides inducing a ground-state dipole of 4.1 D/residue (pointing from the N- to the C-terminus), and type II with “trans” amides has a dipole of 1.5 D/residue (pointing from the C- to the N-terminus). Therefore, conformational changes back and forth from type I to type II polyproline helices switch the direction of their polarization,27 which is a unique feature of these biomolecular electrets. The dipole-generated electric fields in the proximity of protein helices amounts to about to , which proves important for ET and ion-transport processes in living organisms.28–31 In synthetic polypeptide helices, the intrinsic dipoles rectify the directionality of CT.32–37 The broadband gap [or highest occupied molecular orbital (HOMO)–lowest unoccupied molecular orbital (LUMO) gap] of proteins, however, along with the inability to inject charges in them without chemically cleaving their backbones,38–40 considerably limits their utility as electronic materials. Proteins and polypeptides (composed of native -amino acids and their derivatives) mediate ET solely via tunneling, the efficiency of which is limited to about 2 nm.41–46 Conversely, arrays of cofactors and redox-active residues, such as tryptophan or tyrosine, make it possible to attain long-range ET.47,48 That is, multiple short and efficient “electron hopping” steps along arrays of redox moieties allow ET to exceed the 2-nm tunneling limit.48–52 Deoxyribonucleic acid (DNA) strands represent a class of biopolymers that can mediate CT at distances exceeding 2 nm.53–57 Short efficient tunneling steps between closely situated bases, with relatively negative reduction potentials of their radical cations, mediate hole hopping. The lack of detectable distance dependence of the CT rates (for a donor–acceptor separation larger than about 1 nm) is an indication for the hopping mechanism. (In a case of tunneling, the CT rates, , fall off exponentially with donor–acceptor distance, , i.e., , while in a case of hopping, they fall off as an inverse of the number of hopping sites, , i.e., .)49–51 As an alternative to DNA, peptide nucleic acid (PNA) strands present certain advantages for long-range hole hopping.58–61 The smaller helical twist of PNA, in comparison with DNA, improves the electronic coupling between neighboring bases.58 Furthermore, PNA derivatives are not acids (despite the name) and they do not contain ionic charges along their backbones. Instead, PNA backbones comprise primary and secondary amides resulting in the intrinsic electric dipole reported for single-stranded structures.62 The double-stranded PNA, however, has certain conformational rigidity and an advantage for CT over the single-stranded one. To attain permanent intrinsic dipoles in double-stranded structures, the PNA chains have to assemble in a parallel manner. Conversely, the antiparallel double-stranded PNA is slightly more stable than the parallel one.63,64 Considering the electronic features of the various natural macromolecular conjugates, we undertake a bioinspired approach to the development of molecular electrets that can mediate CT (Fig. 1).3 Our de novo designs are based on derivatives of anthranilic acid, which is an aromatic β-amino acid. Similar to protein - and -helices, Aa has ordered arrangements of amide and hydrogen bonds that result in permanent dipoles in the order of 4 to 5 D/residue pointing from the N- to the C-terminus (Fig. 1).2,9 Unlike protein helices, however, the aromatic residues can provide sites for charge hopping along the Aa backbones. In DNA and PNA, the bases providing the charge hopping sites are electronically coupled via noncovalent face-to-face -stacking. In Aa, the aromatic moieties compose the backbones and the partially -conjugated covalent amide linkers ensure considerable overlap between the frontier orbitals on the neighboring residues.9,65 Furthermore, the ability to adjust the electronic properties of each residue by synthetically altering the distal substituents ( and , Fig. 1) provides an incomparable unique means for tuning the CT mechanism from tunneling to hole hopping and to electron hopping. 3.Principles of Biodiversity Extended to Electronic MaterialsThe diversity of life on Earth is made possible through a variety of protein structures and functionalities that stem from only 20 common native -amino acids. The sequence of amino acids in a protein chain directs the folds toward the preferred secondary conformations, tertiary structures, and quaternary self-assemblies that result in various protein functionalities. Cofactors and “noncommon” proteinogenic amino acids, such as selenocysteine and pyrrolysine, may appear as the centerpieces of observed protein activity. It is the whole protein microenvironment, however, with nanometer scale structural features that tunes the vital functionalities. Overall, the properties of the single side chain of each residue and its place in the protein sequence determine the emergence of the structure–function relationships at a macromolecular level. That is, altering the sequences comprising a few “common” residues that differ only by a single side chain leads to countless functionalities. Can electronic and photonic material achieve the same diversity by employing similar principles? The bioinspired molecular electrets are composed of -amino acids, i.e., Aa residues (Fig. 1). Each Aa residue has two side chains, and (Fig. 1), that can be synthetically modified (Fig. 3). The availability for variations at two sites, and , provides a certain advantage over the native -amino acids that contain only a single side chain each. Fig. 3Aa residues, with alkyl-terminated C- and N-termini, used for spectroscopic and electrochemical studies. 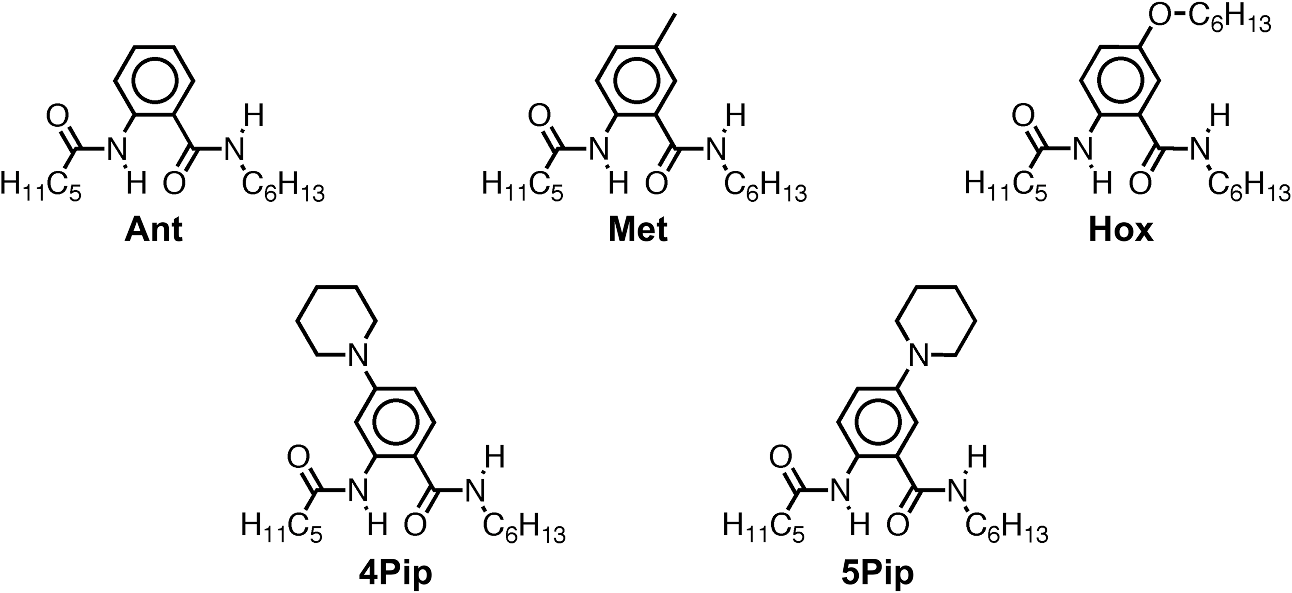 For hole-transducing molecular electrets, we prepared Aa residues with EDGs as the and substituents (Fig. 3). Replacing the hydrogen at with a strong EDG, such as dialkylamine, causes about 1 V negative shift in the reduction potentials [compare 5Pip and Ant, Figs. 3 and 4(b)]. This finding is in an agreement with the 1-eV elevation of the energy levels of the HOMOs induced by similar dialkylamine substituents,9 i.e., such EDGs considerably improve the capabilities of the Aa residues to mediate hole transduction. The electrochemical reduction potentials of singly oxidized organic species, i.e., of , correlate with their ionization energies and hence, with the energy levels of their HOMOs.66–68 Alkyl and alkyloxy substituents that are weaker EDGs than the amines also cause negative shifts in the reduction potential of Aa but not as large as the one observed for [Fig. 4(b)]. Fig. 4Photophysical and electrochemical properties of different Aa residues (Fig. 3). (a) UV/visible absorption and fluorescence spectra of 5Pip for dichloromethane (DCM) (for fluorescence, ). The wavelength, , at the crossing point between the intensity-normalized spectra provides an estimate for the zero-to-zero energy, , which represents the highest occupied molecular orbital (HOMO)–lowest unoccupied molecular orbital (LUMO) optical gap.5,65,69–71 Inset: cyclic voltammogram of 5Pip for DCM in the presence of 200 mM tetrabutylammonium hexafluorophosphate (). (b) Reduction potentials of singly oxidized Aa residues, (), obtained from cyclic voltammetry for solvents with different polarity (as represented by the solvent static relative dielectric constants, ). The data points (the circles) represent for four neat solvents, obtained from extrapolation to zero electrolyte concentration.72,73 The dashed lines are least-square data fits using , as expected from the Born solvation energy.74 (c) Zero-to-zero energy of Aa residues. The circles are data points for four different solvents and dotted lines are linear data fits. [For (b) and (c), the four solvents with different are DCM, benzonitrile, acetonitrile, and propylene carbonate.] 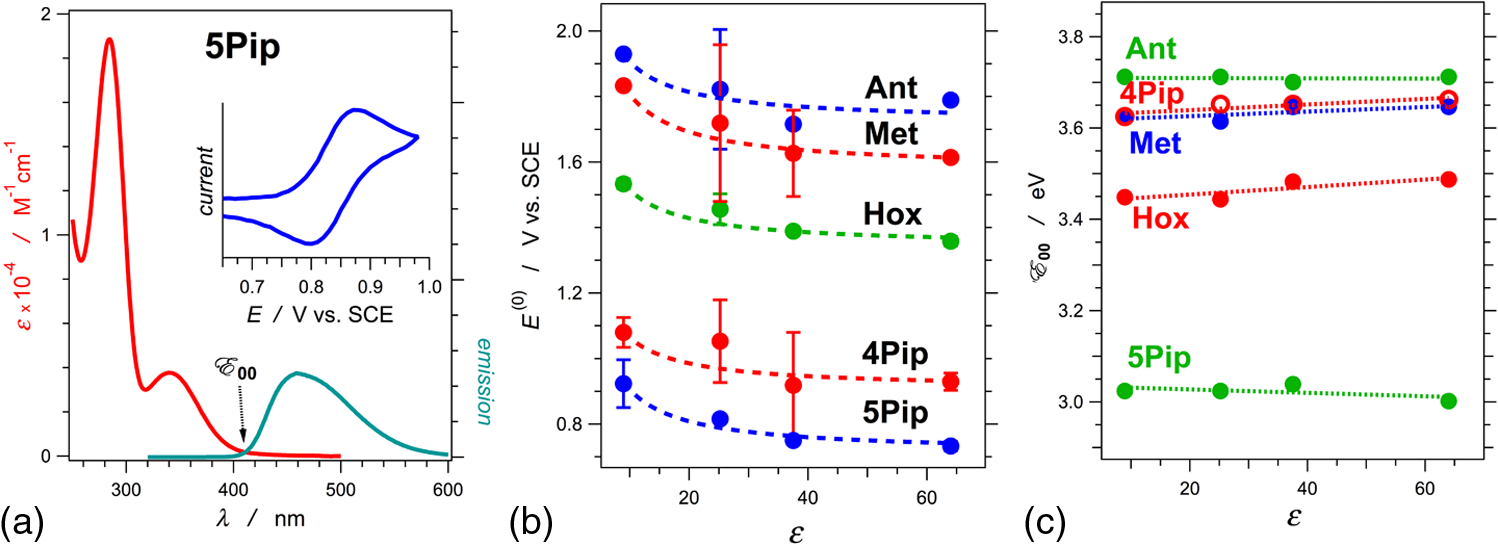 Another feature of the electronic properties of the Aa residues is the significance of the exact position of the substituent; i.e., it is important not only what the substituent is but also where it is in the aromatic ring, i.e., versus . Moving dialkylamine from the to the position causes about a 0.2-V positive shift in the reduction potential [Fig. 4(b)], making the Aa residue a slightly worse mediator of hole transduction. The optical properties of the residues also manifest a strong dependence not only on the type of substituent but also on its position [Fig. 4(c)]. Thus, a single substituent for the two side chains, and , of the Aa residues provide a means for tripling the diversity in properties compared to native amino acids with single side chains. For an electron-donating or electron-withdrawing substituent, G, the three possible Aa residues, , and , would have different electronic properties. This trend demonstrates one of the key advantages of bioinspired approaches versus the biomimetic and biological ones.3 An important feature revealed by the experimental findings is the difference in the extent with which the substituents affect the electrochemical and optical properties of the Aa residues. The reduction potentials of , which correlate with the ionization energy of the residues, pronouncedly depend on the electron-donating strength of the substituents. The exact position of the substituents further tunes the ionization energy but it does not dominate the observed trend. That is, the negative shift in the reduction potential, , follows the electron-donating strength of the substituent, [Fig. 4(b)]. The optical properties of the Aa residues, on the other hand, manifest a considerably stronger sensitivity to the position of the substituents than to their electron-donating strength or -conjugating propensity. For the residues with substituents at position 5, Met, Hox and 5Pip (Fig. 3), the zero-to-zero energy, (which is equivalent to optical band gap for molecular spectroscopy) decreases as alters from H to , , and [Fig. 4(c)]. This trend follows the -conjugating propensity of the substituents. Shifting the piperidine substituent from position 5 to 4 (i.e., 5Pip to 4Pip), increases by about 0.6 eV, making it equivalent to of Met [Fig. 4(c)]. Indeed, these trends are based on examining five conjugates, and their universality is still to be tested by expanding the set of Aa residues. Nevertheless, they reveal key design considerations for electronic and photonic organic materials. Inducing CT rectification is the most important electronic feature of the molecular electrets. The intrinsic dipoles remove the degeneracy between the frontier orbitals on neighboring Aa residues resulting in “cascade” energy configurations for driving hole transfer or ET.9 As the length of the molecular electrets increases above 10 nm or so, the energy of each of the frontier orbitals (e.g., HOMOs and LUMOs) should asymptotically approach constant values toward one of the termini due to dipole-induced charge displacement, making the “cascade” CT profiles nonlinear. Once charge carriers are injected in a sequence of Aa residues, the intrinsic dipoles can ensure a preference for hole transfer toward the N-termini along the HOMOs of the residues, and for ET toward the C-termini along the LUMOs. The capability of Aa conjugates to act as molecular rectifiers (or diodes) can prove immensely beneficial for enhancing forward ET or hole transfer and suppressing undesired charge recombination. These rectifying properties can be attained with homomeric molecular electrets based on sequences of the same Aa residue selected for its ionization energy or electron affinity. Conversely, at donor–acceptor interfaces, diversity in the electronic properties of the Aa residues can significantly aid the efficacy of the initial processes that convert the absorbed light energy into charge carriers. Coulomb trapping of charge-separation states at donor–acceptor interfaces is a principal source of losses in organic photovoltaic and other electronic devices.14,75–83 It also imposes a need for larger driving forces than that required by the fundamental limits (decreasing, for example, the open circuit voltages, ) in order to provide excess energy for the generated charges to escape the traps.14,84–87 Even in the presence of favorably oriented dipoles, these trapping Coulomb interactions can extend over a nanometer or two from the donor–acceptor interfaces, and the depth of the Coulomb traps can be a few hundred millielectronvolts. Hence, because of the presence of such traps for the electret dipoles to exert a dominant effect, the electron or the hole has to migrate a couple of nanometers away from the countercharge at the donor–acceptor interface. Because the increment of Aa polyamides is about , the first two-to-four residues from the donor–acceptor interface are most important for eliminating undesired Coulomb trapping. Considering, for example, hole hopping (i.e., on-resonance hole transfer) along Aa sequences, selecting the first few residues attached to the electron acceptor to have slightly higher ionization energies than the residues in the rest of the polyamide chain can decrease the depth of the Coulomb traps and even completely eliminate them. Molecular electrets with widely diverse sequences will allow for implementing this beneficial CT feature to different electronic materials and energy-conversion systems. In parallel with proteins and their amino-acid building blocks, such diversity in molecular electrets can originate from a few Aa residues with different ionization energies and electron affinities. 4.Experimental Methods4.1.MaterialsThe synthetic starting materials and reagents were purchased from TCI America or Sigma-Aldrich. The solvents (reagent, high-performance-liquid-chromatography, and spectroscopic grade) were obtained from Fisher Scientific. The deuterated solvents were purchased from Cambridge Isotope. The Aa residues (Fig. 3) are prepared from derivatives of 2-nitrobensoic acid with different and substituents (Fig. 1). A general synthetic route includes:1,2 (1) attaching the desired and group, (2) coupling an alkylamine to the C-terminal carboxylate, (3) selectively reducing the nitro group to an amine, and (4) coupling an aliphatic carboxylic acid to the thus formed N-terminal amine. For Ant and Met, the precursors (2-nitrobenzoic acid and 5-methyl-2-nitrobenzoic acid) are commercially available, allowing for the elimination of step 1. All reactions are performed under a nitrogen atmosphere. To prevent undesired oxidation, step 4 is carried out immediately after step 3, without isolating the amine. The combined yields for steps 3 and 4 range between about 10% and 50% regardless of whether tin (II)88 or zinc/ammonium formate89 is used for the reduction of the nitrogroup. All compounds are confirmed using NMR spectroscopy and high resolution mass spectrometry (HRMS). Chemical shifts are reported in parts per million relative to (, ; , ). Data for NMR are reported as follows: chemical shift, integration, multiplicity (, , , , , ), integration, and coupling constants. All NMR spectra were recorded with complete proton decoupling. 4.1.1.Hexyl-N-hexanoylanthranilamide (Ant)The 3-mmol 2-nitrobenzoic acid and 6 mmol -hydroxysuccinimide were dissolved in 10 ml -dimethylformamide (DMF). The solution was chilled on an ice bath and 9 mmol of an activation agent, -diisopropylcarbodiimide, was added dropwise. After stirring for 2 h at 0°C, 9 mmol of 1-aminohexane was added dropwise. The mixture was stirred for 30 more min at 0°C, allowed to warm up to room temperature and stirred until the completion of the reaction: the consumption of the nitrobenzoic acid was monitored using thin-layer chromatography (TLC). The reaction mixture was diluted with dichloromethane (DCM), washed with 5% HCl and MilliQ water, dried with anhydrous , concentrated in vacu and purified chromatographically to produce 550-mg white solid of -hexyl-2-nitrobenzamide (2.2 mmol, 73% yield): -NMR (400 MHz, ) : 7.90 (1 H, dd, , ), 7.55 (1 H, td, , ), 7.46 (1 H, td, , ), 7.37 (1 H, dd, , ), 6.40 (1 H, t, ), 3.28 (2 H, td, , ), 1.51 (2 H, p, ), 1.26 (6 H, m), 0.84 (3 H, t, ); -NMR (400 MHz, ) : 166.61, 146.49, 133.69, 133.24, 130.30, 128.84, 124.44, 40.36, 31.58, 29.22, 26.67, 22.67, 14.14; HRMS m/z calculated for 251.1396, found 251.1393 . -hexyl-2-nitrobenzamide (1 mmol) and (sixfold molar excess) were suspended in 2 ml 1,2-dimethoxyethane and purged with nitrogen. The mixture was refluxed and the progress of the reaction was monitored with TLC. After complete reduction of the nitro group (usually about 2 to 5 h), the solvent was removed under reduced pressure, the reaction solid was suspended in 3 ml DMF under nitrogen, and 1.5 mmol hexanoic anhydride was added dropwise, followed by a slow addition of 1.5 mmol triethylamine, and the mixture was stirred for 4 h. The reaction mixture was dissolved in 50 ml DCM, washed with a saturated aqueous solution of , and dried over anhydrous . Purification using flash chromatography resulted in 30 mg of white compound, Ant (0.9 mmol, 9% yield): -NMR (400 MHz, ) : 11.00 (1 H, s), 8.56 (1 H, d, ), 7.42 (2 H, m), 7.01 (1 H, s), 6.31 (1 H, s), 3.40 (2 H, q, ), 2.36 (2 H, t, ), 1.70 (2 H, p, ), 1.60 (2 H, p, ), 1.3 (10 H, m), 0.88 (6 H, t, ); -NMR (400 MHz, ) : 172.46, 169.21, 139.75, 132.58, 126.51, 122.72, 121.72, 120.83, 40.27, 38.69, 31.68, 31.59, 29.86, 26.87, 25.48, 22.78, 22.61, 14.22, 14.15; HRMS m/z calculated for 341.2199, found 341.2196 . 4.1.2.Hexyl-N-hexanoyl-5-methylanthranilamide (Met)Starting with 5-methyl-2-nitrobenzoic acid and using the procedures for -hexyl-2-nitrobenzamide, afforded -hexyl-5-methyl-2-nitrobenzamide (21% yield): -NMR (400 MHz, ) : 7.89 (1 H, d, ), 7.27 (1 H, dd, , ), 7.21 (1 H, d, ), 6.02 (1 H, s), 3.35 (2 H, q, ), 2.40 (3 H, s), 1.56 (2 H, p, ), 1.3 (6 H, m), 0.86 (3 H, t, );-NMR (400 MHz, ) : 166.96, 145.45, 144.08, 133.52, 130.73, 129.46, 124.72, 40.44, 31.64, 29.36, 26.76, 22.73, 21.54, 14.19; HRMS m/z calculated for 265.1547, found 265.1556 . From -hexyl-5-methyl-2-nitrobenzamide using the procedure for Ant, afforded Met with 25% yield: -NMR (400 MHz, ) : 10.85 (1 H, s), 8.44 (1 H, dd, , ), 7.23 (1 H, d, ), 7.18 (1 H, d, ), 6.24 (1 H, s), 3.39 (2 H, q, ), 2.35 (2 H, t, ), 2.29 (3 H, s), 1.70 (2 H, p, ), 1.60 (2 H, p, ), 1.3 (10 H, m), 0.88 (3 H, t, ), 0.87 (3 H, t, ); -NMR (400 MHz, ) : 172.44, 169.29, 137.03, 133.07, 132.35, 126.95, 121.80, 121.09, 40.25, 38.56, 34.28, 31.67, 31.56, 29.64, 26.86, 25.50, 22.74, 22.57, 14.19, 14.11; HRMS m/z calculated for 355.2356, found 355.2371 . 4.1.3.Hexyl-N-hexanoyl-4-(piperidin-N-yl)anthranilamide (4Pip)The 4-mmol 4-fluoro-2-nitrobenzoic acid was suspended in 2 ml piperidine and refluxed overnight. The reaction solution turned from clear to dark orange, indicative of the formation of 2-nitro-4-(piperidin--yl)benzoic acid. After cooling, the reaction solution was diluted with 100 ml DCM and sequentially washed with 1M HCl and brine. The organic layer was collected, dried over , and concentrated in vacuo to produce 980 mg yellow powder (3.9 mmol, 97%) of 2-nitro-4-(piperidin--yl)benzoic acid:-NMR (400 MHz, ) : 7.84 (1 H, d, ), 6.87 (2 H, m), 3.38 (4 H, m), 1.67 (6 H, s); -NMR (400 MHz, ) : 168.94, 154.18, 153.38, 133.58, 114.46, 107.88, 48.61, 25.35, 24.28; HRMS m/z calculated for 268.1292, found 268.1304 . Using the thus obtained precursor for 4Pip, i.e., 2-nitro-4-(piperidin--yl)benzoic acid, with the procedure for -hexyl-2-nitrobenzamide, afforded -hexyl-2-nitro-4-(piperidin--yl)benzamide (yellow solid) with 75% yield: -NMR (400 MHz, ) : 7.17 (2 H, m), 6.85 (1 H, dd, , ), 5.46 (1 H, t, ), 3.20 (6 H, m), 1.59 (6 H, m), 1.46 (2 H, p, ), 1.22 (6 H, m), 0.81 (3 H, t, );-NMR (400 MHz, ) : 166.62, 152.28, 148.61, 129.51, 120.85, 117.79, 109.56, 48.96, 40.14, 31.54, 29.26, 26.62, 25.22, 24.08, 22.59, 14.06; HRMS m/z calculated for 334.2125, found 334.2139 . Using the procedure for Ant with -hexyl-2-nitro-4-(piperidin--yl)benzamide, afforded 4Pip with 21% yield: -NMR (400 MHz, ) : 11.73 (1 H, s), 8.28 (1 H, d, ), 7.29 (1 H, d, ), 6.39 (2 H, m), 3.31 (2 H, q, ), 3.22 (4 H, m), 2.33 (2 H, t, ), 1.66 (2 H, p, ), 1.55 (8 H, m), 1.27 (10 H, m), 0.83 (6 H, tt, , );-NMR (400 MHz, ) : 172.58, 169.20, 154.20, 142.05, 127.82, 108.41, 108.30, 105.81, 48.78, 39.94, 38.79, 31.62, 31.46, 29.72, 26.81, 25.50, 25.30, 24.46, 22.67, 22.53, 14.11, 14.03; HRMS m/z calculated for 401.3037, found 401.3021 . 4.1.4.N-hexanoyl-5-(piperidin-N-yl)anthranilamide (5Pip)Starting with 4-fluoro-2-nitrobenzoic acid, 5Pip was prepared in four steps using a procedure similar to the one for 4Pip, as we have previously reported.1 4.1.5.Hexyl-N-hexanoyl-5-hexyloxyanthranilamide (Hox)The 5-mmol 5-hydroxy-2-nitrobenzoic acid, 10 mmol , and 11 mmol 1-iodohexane were suspended in 100-ml anhydrous -dimethylacetamide and flushed with nitrogen. The reaction mixture immediately turned yellow due to the deprotonating of the hydroxyl group under the basic conditions (forming the para-nitrophenolate ion derivative of the starting material). The mixture was kept at 150°C for 3 h. Upon cooling, the reaction mixture was suspended in DCM, washed with acidic and basic aqueous solutions, and dried over anhydrous to produce a slightly yellow oil. Purification using flash chromatography afforded 1.7 g (4.8 mmol, 96% yield) of hexyl-5-hexyloxy-2-nitrobenzoate: -NMR (400 MHz, ) : 7.93 (1 H, d, ), 6.97 (1 H, d, ), 6.86 (1 H, dd, , ), 4.27 (2 H, t, ), 3.99 (2 H, t, ), 1.75 (2 H, p, ), 1.66 (2 H, p, ), 1.40 (2 H, m), 1.3 (10 H, m), 0.83 (6 H, tt, ); -NMR (400 MHz, ) : 166.27, 163.10, 139.79, 131.63, 126.66, 115.84, 114.69, 69.28, 66.72, 31.51, 31.45, 28.91, 28.31, 25.60, 25.56, 22.61, 22.58, 14.03; HRMS m/z calculated for 352.2118, found 352.2118 . Hexyl-5-hexyloxy-2-nitrobenzoate (930 mg, 2.6 mmol) was dissolved in 2 ml ethanol and while stirring, 1 ml of 3 M KOH in ethanol was added dropwise. The basified solution was heated to 60°C. The progress of the reaction was monitored using TLC. After the complete consumption of the starting material, the reaction solution was allowed to cool to room temperature and quenched by slowly adding it to a mixture of DCM and 5% aqueous HCl. The organic phase was collected, washed with MilliQ water, dried over anhydrous , and concentrated in vacuo to produce white solid (640 mg, 2.4 mmol, 92%) of 5-hexyloxy-2-nitrobenzoic acid:-NMR (400 MHz, ) : 8.01 (1 H, d, ), 7.13 (1 H, d, ), 7.02 (1 H, dd, , ), 4.05 (2 H, t, ), 1.81 (2 H, p, ), 1.45 (2 H, p, ), 1.32 (4 H, m), 0.89 (3 H, t, ); -NMR (400 MHz, ) : 170.89, 163.12, 140.09, 130.38, 126.81, 116.74, 114.92, 69.49, 31.60, 29.00, 25.69, 22.71, 14.15; HRMS m/z calculated for 268.1179, found 268.1192 . Under nitrogen, 5-hexyloxy-2-nitrobenzoic acid (424 mg, 1.2 mmol), ammonium formate (760 mg, 12 mmol) and zinc dust (434 mg, 6.7 mmol) were suspended in 4 ml 1,2-dimethoxyethane and stirred for 6 h. The reaction mixture was filtered and the filtrate was diluted with DCM then washed in 1% HCl and MilliQ water. The solvent was removed in vacuo and 2 ml of DMF was added under nitrogen. Sequentially, hexanoic anhydride (0.23 ml, 1 mmol) and (0.14 ml, 1 mmol) were added dropwise and the mixture was stirred at room temperature for 2 h. The reaction mixture was diluted with 50 ml ethyl acetate, washed with 5% HCl and MilliQ water, dried over , and concentrated in vacuo. Purification using flash chromatography afforded 210 mg white solid (0.48 mmol, 40% yield) of Hox: -NMR (400 MHz, ) : 10.56 (1 H, s), 8.28 (1 H, d, ), 6.91 (1 H, d, ), 6.86 (1 H, dd, , ), 6.79 (1 H, t, ), 3.84 (2 H, t, ), 3.32 (2 H, q, ), 2.28 (2 H, t, ), 1.65 (4 H, m), 1.55 (2 H, p, ), 1.3 (16 H, m), 0.84 (9 H, t, ); -NMR (400 MHz, ) : 172.15, 168.88, 154.44, 132.19, 123.13, 122.86, 117.11, 113.52, 68.57, 40.21, 38.35, 31.70, 31.63, 31.53, 29.53, 29.35, 26.83, 25.80, 25.48, 22.70, 22.52, 14.13, 14.05; HRMS m/z calculated for 441.3088, found 441.3109 . 4.2.Methods4.2.1.UV/visible absorption and emission spectroscopyThe ground-state absorption spectra were recorded in a transmission mode using a JASCO V-670 spectrophotometer (Tokyo, Japan); and the emission spectra were collected with a FluoroLog-3 spectrofluorometer (Horiba-Jobin-Yvon, Edison, New Jersey) as previously reported.90–96 4.2.2.Electrochemical measurementsCyclic voltammetry (CV) was conducted using Reference 600™ Potentiostat/Galvanostat/ZRA (Gamry Instruments, Pennsylvania), equipped with a three-electrode cell, as previously described.72,73 The half-wave potentials, , were determined from the midpoints between the cathodic and anodic peak potentials for reversible oxidation and from the inflection points of the anodic waves for irreversible oxidation. For each residue, CV was carried out for different solvents at different electrolyte concentrations (tetrabutylammonium hexafluorophosphate was used for the electrolyte with concentration varying between 25 to 200 mM). At least three voltammograms () were recorded for each sample condition (Aa residue, solvent and electrolyte concentration). From the dependence of on the electrolyte concentration, the potentials for neat solvents were estimated from extrapolations to zero73 and presented in Fig. 4(b). 5.ConclusionsInducing rectification and managing charge traps (and potential barriers along CT pathways) represent only a couple of examples of the promising features of molecular electrets. Sets of non-native Aa residues, therefore, can serve as principal tools for the development of widely diverse molecular electrets and provide venues for bottom-up designs of functional electronic and photonic materials for energy applications. AcknowledgmentsFunding for this work was provided by the USA National Science Foundation (grants CHE 1465284, CBET 0935995 and CBET 0923408, as well as IGERT DGE 0903667 for J. M. L); and by the UCR Office of Research and Economic Development for their FY14-15 Proof of Concept Award (V. I. V.) and Collaborative Seed Grant (B. A. and V. I. V.) ReferencesD. Bao et al.,
“Dipole-mediated rectification of intramolecular photoinduced charge separation and charge recombination,”
J. Am. Chem. Soc., 136 12966
–12973
(2014). http://dx.doi.org/10.1021/ja505618n JACSAT 0002-7863 Google Scholar
B. Xia et al.,
“Anthranilamides as bioinspired molecular electrets: experimental evidence for a permanent ground-state electric dipole moment,”
J. Org. Chem., 78 1994
–2004
(2013). http://dx.doi.org/10.1021/jo301942g JOCEAH 0022-3263 Google Scholar
V. I. Vullev,
“From biomimesis to bioinspiration: what’s the benefit for solar energy conversion applications?,”
J. Phys. Chem. Lett., 2 503
–508
(2011). http://dx.doi.org/10.1021/jz1016069 1948-7185 Google Scholar
N. Camaioni and R. Po,
“Pushing the envelope of the intrinsic limitation of organic solar cells,”
J. Phys. Chem. Lett., 4 1821
–1828
(2013). http://dx.doi.org/10.1021/jz400374p 1948-7185 Google Scholar
S. Guo et al.,
“Photoinduced electron transfer between pyridine coated cadmium selenide quantum dots and single sheet graphene,”
Adv. Funct. Mater., 23 5199
–5211
(2013). http://dx.doi.org/10.1002/adfm.201203652 AFMDC6 1616-3028 Google Scholar
H. Lu et al.,
“Pyridine-coated lead sulfide quantum dots for polymer hybrid photovoltaic devices,”
Adv. Sci. Lett., 3 101
–109
(2010). http://dx.doi.org/10.1166/asl.2010.1110 1936-6612 Google Scholar
W. Wang et al.,
“Hybrid low resistance ultracapacitor electrodes based on 1-pyrene butyric acid functionalized centimeter-scale graphene sheets,”
J. Nanosci. Nanotechnol., 12 6913
–6920
(2012). http://dx.doi.org/10.1166/jnn.2012.6507 JNNOAR 1533-4880 Google Scholar
J. Lin et al.,
“Supercapacitors based on pillared graphene nanostructures,”
J. Nanosci. Nanotechnol., 12 1770
–1775
(2012). http://dx.doi.org/10.1166/jnn.2012.5198 JNNOAR 1533-4880 Google Scholar
M. K. Ashraf et al.,
“Theoretical design of bioinspired macromolecular electrets based on anthranilamide derivatives,”
Biotechnol. Prog., 25 915
–922
(2009). http://dx.doi.org/10.1002/btpr.v25:4 BIPRET 8756-7938 Google Scholar
H. D. Sikes et al.,
“Rapid electron tunneling through oligophenylenevinylene bridges,”
Science, 291 1519
–1523
(2001). http://dx.doi.org/10.1126/science.1055745 SCIEAS 0036-8075 Google Scholar
S. Y. Sayed et al.,
“Charge transport in molecular electronic junctions: Compression of the molecular tunnel barrier in the strong coupling regime,”
Proc. Natl Acad. Sci. USA, 109 11498
–11503
(2012). http://dx.doi.org/10.1073/pnas.1201557109 PNASA6 0027-8424 Google Scholar
J. R. Heath and M. A. Ratner,
“Molecular electronics,”
Phys. Today, 56 43
–49
(2003). http://dx.doi.org/10.1063/1.1583533 PHTOAD 0031-9228 Google Scholar
J. A. Christians and P. V. Kamat,
“Trap and transfer. Two-step hole injection across the interface in solid-state solar cells,”
ACS Nano, 7 7967
–7974
(2013). http://dx.doi.org/10.1021/nn403058f 1936-0851 Google Scholar
X. Y. Zhu, Q. Yang and M. Muntwiler,
“Charge-transfer excitons at organic semiconductor surfaces and interfaces,”
Acc. Chem. Res., 42 1779
–1787
(2009). http://dx.doi.org/10.1021/ar800269u ACHRE4 0001-4842 Google Scholar
S. Upadhyayula et al.,
“Coatings of polyethylene glycol for suppressing adhesion between solid microspheres and flat surfaces,”
Langmuir, 28 5059
–5069
(2012). http://dx.doi.org/10.1021/la300545v LANGD5 0743-7463 Google Scholar
J. Wan et al.,
“Surface-bound proteins with preserved functionality,”
Ann. Biomed. Eng., 37 1190
–1205
(2009). http://dx.doi.org/10.1007/s10439-009-9673-6 ABMECF 0090-6964 Google Scholar
B. Millare et al.,
“Dependence of the quality of adhesion between polydimethyl siloxane and glass surfaces on the conditions of treatment with oxygen plasma,”
Langmuir, 24 13218
–13224
(2008). http://dx.doi.org/10.1021/la801965s LANGD5 0743-7463 Google Scholar
W. J. Jasper et al.,
“Degradation processes in corona-charged electret filter-media with exposure to ethyl benzene,”
J. Eng. Fibers Fabrics, 2
(4), 19
–24
(2007). JEFFBY 1558-9250 Google Scholar
G. M. Sessler,
“Electrets: recent developments,”
J. Electrostat., 51 137
–145
(2001). http://dx.doi.org/10.1016/S0304-3886(01)00091-2 JOELDH 0304-3886 Google Scholar
L. S. McCarty and G. M. Whitesides,
“Electrostatic charging due to separation of ions at interfaces: contact electrification of ionic electrets,”
Angew. Chem. Int. Ed., 47 2188
–2207
(2008). http://dx.doi.org/10.1002/(ISSN)1521-3773 ACIEF5 1433-7851 Google Scholar
R. Kressmann, G. M. Sessler and P. Gunther,
“Space-charge electrets,”
IEEE Trans. Dielect. Electr. Insul., 3 607
–623
(1996). http://dx.doi.org/10.1109/94.544184 ITDIES 1070-9878 Google Scholar
R. P. Feynman et al., The Feynman Lectures on Physics: Mainly Electromagnetism and Matter, 2 Addison-Wesley Publishing Company, Boston, MA
(1977). Google Scholar
S. Upadhyayula et al.,
“Permanent electric dipole moments of carboxyamides in condensed media: what are the limitations of theory and experiment?,”
J. Phys. Chem. B, 115 9473
–9490
(2011). http://dx.doi.org/10.1021/jp2045383 JPCBFK 1520-6106 Google Scholar
W. G. J. Hol, P. T. Van Duijnen and H. J. C. Berendsen,
“The alpha-helix dipole and the properties of proteins,”
Nature, 273 443
–446
(1978). http://dx.doi.org/10.1038/273443a0 NATUAS 0028-0836 Google Scholar
W. G. J. Hol,
“Effects of the alpha-helix dipole upon the functioning and structure of proteins and peptides,”
Adv. Biophys., 19 133
–165
(1985). http://dx.doi.org/10.1016/0065-227X(85)90053-X ADVBAT 0065-227X Google Scholar
A. Wada,
“Dielectric properties of polypeptide solutions. II. Relation between the electric dipole moment and the molecular weight of the alpha-helix,”
J. Chem. Phys., 30 328
–329
(1959). http://dx.doi.org/10.1063/1.1729911 JCPSA6 0021-9606 Google Scholar
Y.-G. K. Shin et al.,
“Distance dependence of electron transfer across peptides with different secondary structures: the role of peptide energetics and electronic coupling,”
J. Am. Chem. Soc., 125 3722
–3732
(2003). http://dx.doi.org/10.1021/ja020358q JACSAT 0002-7863 Google Scholar
D. A. Doyle et al.,
“The structure of the potassium channel: molecular basis of K+ conduction and selectivity,”
Science, 280 69
–77
(1998). http://dx.doi.org/10.1126/science.280.5360.69 SCIEAS 0036-8075 Google Scholar
R. Dutzler et al.,
“X-ray structure of a ClC chloride channel at 3.0 Å reveals the molecular basis of anion selectivity,”
Nature, 415 287
–294
(2002). http://dx.doi.org/10.1038/415287a NATUAS 0028-0836 Google Scholar
X. H. Chen et al.,
“Alpha-helix C-terminus acting as a relay to mediate long-range hole migration in proteins,”
J. Phys. Chem. Lett., 1 1637
–1641
(2010). http://dx.doi.org/10.1021/jz100210t 1948-7185 Google Scholar
S. Tanaka and R. A. Marcus,
“Electron transfer model for the electric field effect on quantum yield of charge separation in bacterial photosynthetic reaction centers,”
J. Phys. Chem. B, 101 5031
–5045
(1997). http://dx.doi.org/10.1021/jp9632854 JPCBFK 1520-6106 Google Scholar
E. Galoppini and M. A. Fox,
“Effect of the electric field generated by the helix dipole on photoinduced intramolecular electron transfer in dichromophoric alpha-helical peptides,”
J. Am. Chem. Soc., 118 2299
–2300
(1996). http://dx.doi.org/10.1021/ja951555a JACSAT 0002-7863 Google Scholar
M. A. Fox and E. Galoppini,
“Electric field effects on electron transfer rates in dichromophoric peptides: the effect of helix unfolding,”
J. Am. Chem. Soc., 119 5277
–5285
(1997). http://dx.doi.org/10.1021/ja963269k JACSAT 0002-7863 Google Scholar
S. Yasutomi et al.,
“A molecular photodiode system that can switch photocurrent direction,”
Science, 304 1944
–1947
(2004). http://dx.doi.org/10.1126/science.1098489 SCIEAS 0036-8075 Google Scholar
L. Garbuio et al.,
“Effect of orientation of the peptide-bridge dipole moment on the properties of fullerene-peptide-radical systems,”
J. Am. Chem. Soc., 134 10628
–10637
(2012). http://dx.doi.org/10.1021/ja303696s JACSAT 0002-7863 Google Scholar
H. S. Mandal and H. B. Kraatz,
“Electron transfer across alpha-helical peptides: potential influence of molecular dynamics,”
Chem. Phys., 326 246
–251
(2006). http://dx.doi.org/10.1016/j.chemphys.2006.01.010 CMPHC2 0301-0104 Google Scholar
C. Shlizerman et al.,
“De novo designed coiled-coil proteins with variable conformations as components of molecular electronic devices,”
J. Am. Chem. Soc., 132 5070
–5076
(2010). http://dx.doi.org/10.1021/ja907902h JACSAT 0002-7863 Google Scholar
J. F. Odonnell and C. K. Mann,
“Controlled-potential oxidation of aliphatic amides,”
J. Electroanal. Chem., 13 157
–162
(1967). http://dx.doi.org/10.1016/0022-0728(67)80108-6 JECHES 0022-0728 Google Scholar
L. G. Fong et al.,
“Nonenzymatic oxidative cleavage of peptide-bonds in apoprotein-B-100,”
J. Lipid Res., 28
(12), 1466
–1477
(1987). JLPRAW 0022-2275 Google Scholar
M. J. Davies,
“Protein and peptide alkoxyl radicals can give rise to C-terminal decarboxylation and backbone cleavage,”
Arch. Biochem. Biophys., 336 163
–172
(1996). http://dx.doi.org/10.1006/abbi.1996.0545 ABBIA4 0003-9861 Google Scholar
D. N. Beratan et al.,
“Electron-tunneling pathways in proteins,”
Science, 258 1740
–1741
(1992). http://dx.doi.org/10.1126/science.1334572 SCIEAS 0036-8075 Google Scholar
H. B. Gray and J. R. Winkler,
“Long-range electron transfer,”
Proc. Natl Acad. Sci. USA, 102 3534
–3539
(2005). http://dx.doi.org/10.1073/pnas.0408029102 PNASA6 0027-8424 Google Scholar
V. I. Vullev and G. JonesII,
“Photoinduced charge transfer in helical polypeptides,”
Res. Chem. Intermed., 28 795
–815
(2002). http://dx.doi.org/10.1163/15685670260469429 RCINEE 0922-6168 Google Scholar
G. JonesII, X. Zhou and V. I. Vullev,
“Photoinduced electron transfer in alpha-helical polypeptides: dependence on conformation and electron donor-acceptor distance,”
Photochem. Photobiol. Sci., 2 1080
–1087
(2003). http://dx.doi.org/10.1039/b306490e PPSHCB 1474-905X Google Scholar
G. JonesII and V. I. Vullev,
“Photoinduced electron transfer between non-native donor-acceptor moieties incorporated in synthetic polypeptide aggregates,”
Org. Lett., 4 4001
–4004
(2002). http://dx.doi.org/10.1021/ol026656+ ORLEF7 1523-7060 Google Scholar
G. JonesII et al.,
“Multistep photoinduced electron transfer in a de novo helix bundle: multimer self-assembly of peptide chains including a chromophore special pair,”
J. Am. Chem. Soc., 122 388
–389
(2000). http://dx.doi.org/10.1021/ja981936z JACSAT 0002-7863 Google Scholar
C. Shih et al.,
“Tryptophan-accelerated electron flow through proteins,”
Science, 320 1760
–1762
(2008). http://dx.doi.org/10.1126/science.1158241 SCIEAS 0036-8075 Google Scholar
J. J. Warren, J. R. Winkler and H. B. Gray,
“Hopping maps for photosynthetic reaction centers,”
Coord. Chem. Rev., 257 165
–170
(2013). http://dx.doi.org/10.1016/j.ccr.2012.07.002 CCHRAM 0010-8545 Google Scholar
J. Jortner et al.,
“Charge transfer and transport in DNA,”
Proc. Natl Acad. Sci. USA, 95 12759
–12765
(1998). http://dx.doi.org/10.1073/pnas.95.22.12759 PNASA6 0027-8424 Google Scholar
K. Kawai et al.,
“Kinetics of weak distance-dependent hole transfer in DNA by adenine-hopping mechanism,”
J. Am. Chem. Soc., 125 6842
–6843
(2003). http://dx.doi.org/10.1021/ja034953j JACSAT 0002-7863 Google Scholar
C. Behrens et al.,
“Weak distance dependence of excess electron transfer in DNA,”
Angew. Chem. Int. Ed., 41 1763
–1766
(2002). http://dx.doi.org/10.1002/(ISSN)1521-3773 ACIEF5 1433-7851 Google Scholar
G. JonesII et al.,
“Photoactive peptides. 6. Photoinduced electron transfer for pyrenesulfonamide conjugates of tryptophan-containing peptides. Mitigation of fluoroprobe behavior in N-terminal labeling experiments,”
Bioorg. Med. Chem. Lett., 5 2385
–2390
(1995). http://dx.doi.org/10.1016/0960-894X(95)00416-Q BMCLE8 0960-894X Google Scholar
F. D. Lewis,
“Distance-dependent electronic interactions across DNA base pairs: charge transport, exciton coupling, and energy transfer,”
Isr. J. Chem., 53 350
–365
(2013). http://dx.doi.org/10.1002/ijch.v53.6/7 ISJCAT 0021-2148 Google Scholar
K. Kawai and T. Majima,
“Hole transfer kinetics of DNA,”
Acc. Chem. Res., 46 2616
–2625
(2013). http://dx.doi.org/10.1021/ar400079s ACHRE4 0001-4842 Google Scholar
N. Renaud et al.,
“Between superexchange and hopping: an intermediate charge-transfer mechanism in poly(A)-poly(T) DNA hairpins,”
J. Am. Chem. Soc., 135 3953
–3963
(2013). http://dx.doi.org/10.1021/ja3113998 JACSAT 0002-7863 Google Scholar
F. D. Lewis et al.,
“Crossover from superexchange to hopping as the mechanism for photoinduced charge transfer in DNA hairpin conjugates,”
J. Am. Chem. Soc., 128 791
–800
(2006). http://dx.doi.org/10.1021/ja0540831 JACSAT 0002-7863 Google Scholar
K. Senthilkumar et al.,
“Absolute rates of hole transfer in DNA,”
J. Am. Chem. Soc., 127 14894
–14903
(2005). http://dx.doi.org/10.1021/ja054257e JACSAT 0002-7863 Google Scholar
R. Venkatramani et al.,
“Nucleic acid charge transfer: black, white and gray,”
Coord. Chem. Rev., 255 635
–648
(2011). http://dx.doi.org/10.1016/j.ccr.2010.12.010 CCHRAM 0010-8545 Google Scholar
E. Hatcher et al.,
“PNA versus DNA: effects of structural fluctuations on electronic structure and hole-transport mechanisms,”
J. Am. Chem. Soc., 130 11752
–11761
(2008). http://dx.doi.org/10.1021/ja802541e JACSAT 0002-7863 Google Scholar
R. Venkatramani et al.,
“Evidence for a near-resonant charge transfer mechanism for double-stranded peptide nucleic acid,”
J. Am. Chem. Soc., 133 62
–72
(2011). http://dx.doi.org/10.1021/ja107622m JACSAT 0002-7863 Google Scholar
E. Wierzbinski et al.,
“The single-molecule conductance and electrochemical electron-transfer rate are related by a power law,”
ACS Nano, 7 5391
–5401
(2013). http://dx.doi.org/10.1021/nn401321k 1936-0851 Google Scholar
M. A. Wolak et al.,
“Electronic structure of self-assembled peptide nucleic acid thin films,”
J. Phys. Chem. C, 115 17123
–17135
(2011). http://dx.doi.org/10.1021/jp201602j 1932-7447 Google Scholar
M. Egholm et al.,
“PNA hybridizes to complementary oligonucleotides obeying the Watson-Crick hydrogen-bonding rules,”
Nature, 365 566
–568
(1993). http://dx.doi.org/10.1038/365566a0 NATUAS 0028-0836 Google Scholar
P. E. Nielsen and G. Haaima,
“Peptide nucleic acid (PNA). A DNA mimic with a pseudopeptide backbone,”
Chem. Soc. Rev., 26 73
–78
(1997). http://dx.doi.org/10.1039/cs9972600073 CSRVBR 0306-0012 Google Scholar
J. Wan et al.,
“Solvent dependence of the charge-transfer properties of a quaterthiophene-anthraquinone dyad,”
J. Photochem. Photobiol. A, 197 364
–374
(2008). http://dx.doi.org/10.1016/j.jphotochem.2008.01.016 JPPCEJ 1010-6030 Google Scholar
H. D. Burrows,
“The relation between standard electrode and ionization potentials of metal ions,”
J. Chem. Educ., 53 365
(1976). http://dx.doi.org/10.1021/ed053p365 JCEDA8 0021-9584 Google Scholar
A. Dhiman et al.,
“A simple correlation of anodic peak potentials of silylarenes and their vertical ionization energies,”
Organometallics, 23 1636
–1638
(2004). http://dx.doi.org/10.1021/om030609j ORGND7 0276-7333 Google Scholar
P. Muller,
“Glossary of terms used in physical organic-chemistry,”
Pure Appl. Chem., 66 1077
–1184
(1994). http://dx.doi.org/10.1351/pac199466051077 PACHAS 0033-4545 Google Scholar
J. Hu et al.,
“Long-lived photogenerated states of -oligothiophene-acridinium dyads have triplet character,”
J. Phys. Chem. A, 113 3096
–3107
(2009). http://dx.doi.org/10.1021/jp810909v JPCAFH 1089-5639 Google Scholar
V. I. Vullev and G. Jones,
“Photoinduced electron transfer in alkanoylpyrene aggregates in conjugated polypeptides,”
Tetrahedr. Lett., 43 8611
–8615
(2002). http://dx.doi.org/10.1016/S0040-4039(02)01895-6 TELEAY 0040-4039 Google Scholar
G. JonesII et al.,
“Photoinduced electron transfer in arylacridinium conjugates in a solid glass matrix,”
J. Phys. Chem. B, 111 6921
–6929
(2007). http://dx.doi.org/10.1021/jp072224a JPCBFK 1520-6106 Google Scholar
D. Bao et al.,
“Electrochemical reduction of quinones: interfacing experiment and theory for defining effective radii of redox moieties,”
J. Phys. Chem. B, 114 14467
–14479
(2010). http://dx.doi.org/10.1021/jp101730e JPCBFK 1520-6106 Google Scholar
D. Bao et al.,
“Electrochemical oxidation of ferrocene: a strong dependence on the concentration of the supporting electrolyte for nonpolar solvents,”
J. Phys. Chem. A, 113 1259
–1267
(2009). http://dx.doi.org/10.1021/jp809105f JPCAFH 1089-5639 Google Scholar
M. Born,
“Volumes and heats of hydration of ions,”
Z. Phys., 1 45
–48
(1920). http://dx.doi.org/10.1007/BF01881023 ZEPYAA 0044-3328 Google Scholar
Q. Yang, M. Muntwiler and X. Y. Zhu,
“Charge transfer excitons and image potential states on organic semiconductor surfaces,”
Phys. Rev. B, 80 115214
(2009). http://dx.doi.org/10.1103/PhysRevB.80.115214 PRBMDO 1098-0121 Google Scholar
I. Lalov, C. Warns and P. Reineker,
“Model of mixed Frenkel and charge-transfer excitons in donor-acceptor molecular crystals: investigation of vibronic spectra,”
New J. Phys., 10 085006
(2008). http://dx.doi.org/10.1088/1367-2630/10/8/085006 NJOPFM 1367-2630 Google Scholar
M. Muntwiler, Q. Yang and X. Y. Zhu,
“Exciton dynamics at interfaces of organic semiconductors,”
J. Electron Spectrosc. Relat. Phenom., 174 116
–124
(2009). http://dx.doi.org/10.1016/j.elspec.2009.02.016 JESRAW 0368-2048 Google Scholar
M. Yokoyama et al.,
“Mechanism of photocarrier generation in polyvinylcarbazole. Mechanism of extrinsic carrier photogeneration. Quenching of exciplex fluorescence by electric field,”
Polym. Prepr. (Am. Chem. Soc., Div. Polym. Chem.), 20
(April), 399
–402
(1979). ACPPAY 0032-3934 Google Scholar
M. Yokoyama et al.,
“Mechanism of extrinsic carrier photogeneration in poly-N-vinylcarbazole. II. Quenching of exciplex fluorescence by electric field,”
J. Chem. Phys., 75 3006
–3011
(1981). http://dx.doi.org/10.1063/1.442392 JCPSA6 0021-9606 Google Scholar
M. Yokoyama, Y. Endo and H. Mikawa,
“Quenching of exciplex fluorescence by an electric field in the solid state,”
Chem. Phys. Lett., 34 597
–600
(1975). http://dx.doi.org/10.1016/0009-2614(75)85571-0 CHPLBC 0009-2614 Google Scholar
J. P. Schmidtke, R. H. Friend and C. Silva,
“Tuning interfacial charge-transfer excitons at polymer-polymer heterojunctions under hydrostatic pressure,”
Phys. Rev. Lett., 100 157401
(2008). http://dx.doi.org/10.1103/PhysRevLett.100.157401 PRLTAO 0031-9007 Google Scholar
P. Peumans and S. R. Forrest,
“Separation of geminate charge-pairs at donor-acceptor interfaces in disordered solids,”
Chem. Phys. Lett., 398 27
–31
(2004). http://dx.doi.org/10.1016/j.cplett.2004.09.030 CHPLBC 0009-2614 Google Scholar
U. Albrecht and H. Baessler,
“Yield of geminate pair dissociation in an energetically random hopping system,”
Chem. Phys. Lett., 235 389
–393
(1995). http://dx.doi.org/10.1016/0009-2614(95)00121-J CHPLBC 0009-2614 Google Scholar
R. D. Pensack and J. B. Asbury,
“Beyond the adiabatic limit: charge photogeneration in organic photovoltaic materials,”
J. Phys. Chem. Lett., 1 2255
–2263
(2010). http://dx.doi.org/10.1021/jz1005225 1948-7185 Google Scholar
J. Guo et al.,
“Charge generation and recombination dynamics in poly(3-hexylthiophene)/fullerene blend films with different regioregularities and morphologies,”
J. Am. Chem. Soc., 132 6154
–6164
(2010). http://dx.doi.org/10.1021/ja100302p JACSAT 0002-7863 Google Scholar
M. Muntwiler et al.,
“Coulomb barrier for charge separation at an organic semiconductor interface,”
Phys. Rev. Lett., 101
(2008). http://dx.doi.org/10.1103/PhysRevLett.101.196403 PRLTAO 0031-9007 Google Scholar
H. Scher and S. Rackovsky,
“Theory of geminate recombination on a lattice,”
J. Chem. Phys., 81 1994
–2009
(1984). http://dx.doi.org/10.1063/1.447822 JCPSA6 0021-9606 Google Scholar
F. D. Bellamy and K. Ou,
“Selective reduction of aromatic nitro compounds with stannous chloride in nonacidic and nonaqueous medium,”
Tetrahedr. Lett., 25 839
–842
(1984). http://dx.doi.org/10.1016/S0040-4039(01)80041-1 TELEAY 0040-4039 Google Scholar
D. C. Gowda, B. Mahesh and S. Gowda,
“Zinc-catalyzed ammonium formate reductions: rapid and selective reduction of aliphatic and aromatic nitro compounds,”
Ind. J. Chem. B, 40B
(1), 75
–77
(2001). IJSBDB 0376-4699 Google Scholar
J. M. Vasquez et al.,
“Fluorescence enhancement of warfarin induced by interaction with beta-cyclodextrin,”
Biotechnol. Prog., 25 906
–914
(2009). http://dx.doi.org/10.1002/btpr.v25:4 BIPRET 8756-7938 Google Scholar
S. Upadhyayula et al.,
“Photoinduced dynamics of a cyanine dye: parallel pathways of non-radiative deactivation involving multiple excited-state twisted transients,”
Chem. Sci., 6 2237
–2251
(2015). http://dx.doi.org/10.1039/C4SC02881C CESCAC 0009-2509 Google Scholar
B. Jung, V. I. Vullev and B. Anvari,
“Revisiting indocyanine green: effects of serum and physiological temperature on absorption and fluorescence characteristics,”
IEEE J. Sel. Top. Quantum Electron., 20
(2), 7000409
(2014). http://dx.doi.org/10.1109/JSTQE.2013.2278674 IJSQEN 1077-260X Google Scholar
V. Nuñez et al.,
“Microfluidic space-domain time-resolved emission spectroscopy of terbium(III) and europium(III) chelates with pyridine-2,6-dicarboxylate,”
Anal. Chem., 85 4567
–4577
(2013). http://dx.doi.org/10.1021/ac400200x ANCHAM 0003-2700 Google Scholar
M. Ghazinejad et al.,
“Non-invasive high-throughput metrology of functionalized graphene sheets,”
Adv. Funct. Mater., 22 4519
–4525
(2012). http://dx.doi.org/10.1002/adfm.201200434 AFMDC6 1616-3028 Google Scholar
B. Xia et al.,
“Amyloid histology stain for rapid bacterial endospore imaging,”
J. Clin. Microbiol., 49 2966
–2975
(2011). http://dx.doi.org/10.1128/JCM.02285-10 JCMIDW 1070-633X Google Scholar
M. S. Thomas et al.,
“Kinetics of bacterial fluorescence staining with 3,3’-diethylthiacyanine,”
Langmuir, 26 9756
–9765
(2010). http://dx.doi.org/10.1021/la1013279 LANGD5 0743-7463 Google Scholar
BiographyJillian M. Larsen is a senior PhD student in bioengineering at the University of California, Riverside (UCR). She received her BS degree in biochemistry from UCR. Her current research is on the design, preparation, and characterization of dipolar organic conjugates. She developed several procedures, including microwave-aided procedures, for the synthesis of anthranilamides and their precursors. Eli M. Espinoza is a PhD student in chemistry at the University of California, Riverside. He received his BS degree from Azusa Pacific University and his MS degree in chemistry from California State University, Los Angeles. His current research is on synthesis, electrochemistry, and spectroscopy of organic conjugates for photonic and electronic materials. He develops procedures for preparing multifunctional polyaromatics with pronounced regioisomer purity. Valentine I. Vullev is an associate professor at the University of California, Riverside. He received his PhD in chemistry from Boston University and carried out postdoctoral work at Harvard University. His research interests are in charge transfer, photonics, organic electronic materials, and molecular engineering. |




